Endocyclic extension of porphyrin π-system in etheno-bridged N-confused tetraphenylporphyrin†
Motoki Toganoh, Tomoyuki Kimura and Hiroyuki Furuta*
Department of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan. E-mail: hfuruta@cstf.kyushu-u.ac.jp; Fax: +81 92-802-2865; Tel: +81 92-802-2865
First published on 16th October 2007
Abstract
Etheno-bridged N-confused tetraphenylporphyrin was synthesized through flipping of the confused pyrrole ring and endocyclic extension of [18]annulenic π-conjugated system was exemplified by the absorption spectra as well as the calculated Kohn–Sham orbitals.
Extension of π-conjugated systems has continued to gain considerable attention, since it often leads to interesting physical properties, which would open up a new area of research in physical organic chemistry and materials science.1 In most cases, the π-system extension in the cyclic compounds is achieved at the periphery of the molecules and examples of endocyclic extension are quite limited, probably due to the difficulty in regioselective chemical functionalization inside the macrocycles.
Porphyrins are well-known π-conjugated macrocyclic compounds and the exocyclic extension of the [18]annulenic π-system has been extensively studied.2,3 Contrastingly, the endocyclic extension is usually considered to be impossible, since no accessible sp2 carbon atoms are left inside the macrocyclic core. Meanwhile, a porphyrin analogue, carbaporphyrin, has a free sp2 carbon atom in its core, and therefore, endocyclic π-system extension is essentially possible.4 Nevertheless, a lack of a general method to introduce substituents into the inner sp2 carbon atom, certainly due to the steric problem, has hindered the progress of research. One fascinating remedy against such a difficulty could be a confusion approach which utilizes the inversion of pyrrole rings.5 In N-confused porphyrins,6 the regioselective introduction of a bulky substituent into the inner sp2 carbon atom was achieved by using the ring flip of the confused pyrrole (Scheme 1).7,8 Through the transformation from N-confused porphyrin (A) to N-fused porphyrin (B), the carbon atom originally placed inside the core moves to the peripheral position. Chemical functionalization of the exposed sp2 carbon atom in B would be easily accomplished to give the functionalized N-fused porphyrin (C) and a further treatment with a nucleophile causes the regeneration of N-confused porphyrin (D), where the functional group is located inside the core. This time, the endocyclic extension of the porphyrin π-system has been achieved by the confusion approach and etheno-bridged N-confused tetraphenylporphyrins (1a and 1b) are successfully synthesized, where the extension of the porphyrin π-system is confirmed by the absorption spectra as well as the calculated Kohn–Sham orbitals.9
 | ||
| Scheme 1 Confusion approach to endocyclic chemical functionalization of porphyrins. | ||
The preparation of the etheno-bridged N-confused tetraphenylporphyrins was carried out as shown in Scheme 2. Starting from N-confused tetraphenylporphyrin (3), the brominated N-fused tetraphenylporphyrin (4) was synthesized in two steps as reported previously.10,11 Then, a trimethylsilylethynyl group was introduced by Pd-catalyzed Stille coupling reaction of 4 with the corresponding tin reagent to give the alkynylated product (5) in 83% yield. Finally, the inversion of the confused pyrrole ring was examined by treatment of 5 with NaOMe or NaOEt in THF. Surprisingly, the isolated product was not the expected N-confused porphyrin having an alkynyl group at the core but the etheno-bridged N-confused tetraphenylporphyrins. While no direct evidence is obtained yet, 1 would be produced by the intramolecular addition reaction of the pyrrolic nitrogen to the alkyne moiety via the ethynylated N-confused tetraphenylporphyrin (6), which is supported by the further experiments described below. Interestingly, the reaction afforded only one type of product (1a or 1b) in good yields and the isomer (2a or 2b) was not detected at all.
 | ||
| Scheme 2 Synthesis of etheno-bridged N-confused porphyrins. | ||
Remarkable selectivity in the production of 1a is commonly observed regardless of the starting compounds (Scheme 3). First, the conversion from 5 to 1a was achieved in a stepwise manner. Deprotection of 5 with tetrabutylammonium fluoride (TBAF) afforded 21-ethynyl N-fused tetraphenylporphyrin (7) in 89% yield. Treatment of 7 with NaOMe gave 1a in 60% yield. While the yield became lower than that of one-pot reaction, the isomer 2a was never detected by TLC and 1H NMR analyses. The plausible intermediate 6 was not obtained. Then, the deprotection was performed after the inversion from N-fused porphyrin to N-confused porphyrin. The triisopropylsilyl derivative 8 was prepared in 78% yield by the Stille coupling reaction of 4 in the same manner as the preparation of 5. Next, 8 was reacted with NaOMe, which gave the interiorly ethynylated N-confused tetraphenylporphyrin (9) in 73% yield. The intramolecular addition reaction from 9 to 10 does not proceed at ambient temperature and thus 9 is stable enough to be isolated. Finally, deprotection of the triisopropylsilyl group was performed with TBAF. In this reaction, similar to the one-pot reaction from 5, only the single isomer (1a) was isolated in 97% yield and 6 was never obtained.
 | ||
| Scheme 3 Stepwise preparation of 1a. | ||
The structural assignment of the etheno-bridged N-confused porphyrins rests on X-ray crystallographic analysis on a single crystal of 1b.12 The ORTEP drawing of 1b is shown in Fig. 1. The etheno-bridged structure between the inner carbon atom of the confused pyrrole and the adjacent pyrrole nitrogen atom, which lies 1.271 Å above the porphyrin mean plane composed of the 24 heavy atoms, is clearly observed. The C1–C2 bond length is 1.337(6) Å, which is a typical length for carbon–carbon double bonds. This bond length indicates that the strain present in the unique [5.7.5] tricyclic ring system is modest.
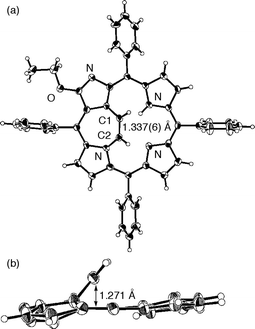 | ||
| Fig. 1 The ORTEP drawing of 1b at the 30% probability level: (a) top view, (b) side view. The phenyl and ethoxy groups are omitted for clarity in (b). | ||
All the spectroscopic data of 1a and 1b are fully consistent with the assigned structures. The MALDI-TOF mass analysis affords the expected values for M+ (1a: m/z 668.2, 1b: m/z 682.0). The signals due to the two olefinic protons inside the macrocycle of 1a appear at δ−0.78 and −0.47 ppm, which couple with each other (3JHH = 7.3 Hz) in the 1H NMR spectrum. Corresponding signals for 1b appear in the same way as those of 1a.
While the reason for the exclusive formation of 1 over 2 is unclear yet, it would be due to a kinetic reason since 2a is suggested to be more stable than 1a by 2.66 kcal mol−1 according to B3LYP/6-311++G**//B3LYP/6-31G** level calculations (Fig. 2). In the optimized structures, no prominent difference is observed between 1a and 2a except for the positions of the inner olefin moiety. For example, the lengths of the inner olefins are 1.336 Å for 1a and 1.341 Å for 2a. Unlike the imino-fused N-confused porphyrin having a similar [5.7.5] tricyclic ring structure,13 the thermal isomerization from 1a to 2a has not been observed yet. When 1a was heated to 180 °C in 1,2-Cl2C6H4 under Ar, only slight decomposition was observed by 1H NMR analysis. Thus, the isomer (2a) is not obtained in spite of its stability inferred from the calculations.
 | ||
| Fig. 2 The optimized structures of (a) 1a and (b) 2a at the B3LYP/6-31G** level. | ||
The endocyclic extension of the porphyrin π-system can be illustrated by photophysical measurements. The absorption spectrum of 1a in CH2Cl2 is shown in Fig. 3 together with that of 3-methoxy N-confused tetraphenylporphyrin (11)10 for comparison. The severe deformation of the Soret band of 1a would imply the unique π-delocalization in the new π-conjugated system. The longest λmax in the Q-band region for 1a appears at 775 nm and that of 11 appears at 706 nm. The observed large red-shift of nearly 70 nm suggests the smaller HOMO–LUMO gap of the etheno-bridged N-confused porphyrins.14 No significant difference from 1a is recognized in the absorption spectrum of 1b (see ESI†).
 | ||
| Fig. 3 Absorption spectra of 1a and 11 in CH2Cl2. | ||
Further evidence of the endocyclic extension of the porphyrin π-system is obtained by theoretical calculations. The calculated orbitals for 1a at the B3LYP/6-31G** level are shown in Fig. 4 together with the calculated HOMO of 11 at the same level. In the HOMO of 1a, a significant contribution of the π-orbital of the etheno-bridge moiety is observed (Fig. 4(a)) and the remaining orbitals are quite similar to those of 11 (Fig. 4(b)). The relationship between the porphyrin π-orbital and the etheno π-orbital is out-of-phase in the HOMO, and one of the in-phase interactions is observed in HOMO−19 (Fig. 4(c)). Contrastingly, no contribution of the etheno part is observed in the LUMO of 1a (Fig. 4(d)). The energy values of HOMO (LUMO) level for 1a and 11 are −0.16647 (−0.8396) and −0.17419 (−0.08318) Hartree, respectively. Hence, the observed smaller HOMO–LUMO gap of 1a than 11 would be attributable to the raising of the HOMO energy level.
 | ||
| Fig. 4 Kohn–Sham orbitals calculated at the B3LYP/6-31G** level. (a) HOMO of 1a, (b) HOMO of 11, (c) HOMO−19 of 1a, and (d) LUMO of 1a. | ||
In conclusion, we have succeeded in extending the porphyrin π-system inside the core using the confusion strategy and the etheno-bridged N-confused porphyrins are synthesized for the first time. The introduction of an etheno-bridge in the N-confused porphyrin would cause the rise of the HOMO energy level, which is illustrated by the absorption spectra and the DFT calculated orbitals. The strategy developed here seems useful for the construction of unique π-conjugated systems and further application is now under investigation.
The present work is partially supported by a Grant-in-Aid for the Global COE Program, “Science for Future Molecular Systems” from the Ministry of Education, Culture, Science, Sports and Technology of Japan.
Notes and references
- (a) R. D. Kennedy, D. Lloyd and H. McNab, J. Chem. Soc., Perkin Trans. 1, 2002, 1601 RSC; (b) M. J. Marsella, Acc. Chem. Res., 2002, 35, 944 CrossRef CAS; (c) M. D. Watson, A. Fechtenkötter and K. Müllen, Chem. Rev., 2001, 101, 1267 CrossRef CAS; (d) R. E. Martin and F. Diederich, Angew. Chem., Int. Ed., 1999, 38, 1350 CrossRef.
- (a) H. L. Anderson, Chem. Commun., 1999, 2323 RSC; (b) D. Kim and A. Osuka, Acc. Chem. Res., 2004, 37, 735 CrossRef CAS; (c) A. K. Burrell, D. L. Officer, P. G. Plieger and D. C. W. Reid, Chem. Rev., 2001, 101, 2751 CrossRef CAS.
- V. S.-Y. Lin, S. G. DiMagno and M. J. Therien, Science, 1994, 264, 1105 CrossRef CAS.
- (a) T. D. Lash, Synlett, 1999, 279; (b) M. Stępień and L. Latos-Grażyński, Acc. Chem. Res., 2005, 38, 88 CrossRef CAS.
- (a) H. Furuta, H. Maeda and A. Osuka, Chem. Commun., 2002, 1795 RSC; (b) J. D. Harvey and C. J. Ziegler, Coord. Chem. Rev., 2003, 247, 1 CrossRef CAS; (c) J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134 CrossRef CAS; (d) T. K. Chandrashekar and S. Venkatraman, Acc. Chem. Res., 2003, 36, 676 CrossRef CAS; (e) A. Ghosh, Angew. Chem., Int. Ed., 2004, 43, 1918 CrossRef CAS; (f) P. L. Chmielewski and L. Latos-Grażyński, Coord. Chem. Rev., 2005, 249, 2510 CrossRef CAS; (g) A. Srinivasan and H. Furuta, Acc. Chem. Res., 2005, 38, 10 CrossRef CAS.
- (a) H. Furuta, T. Asano and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 767 CrossRef CAS; (b) P. J. Chmielewski, L. Latos-Grażyński, K. Rachlewicz and T. Głowiak, Angew. Chem., Int. Ed. Engl., 1994, 33, 779 CrossRef; (c) T. Morimoto, S. Taniguchi, A. Osuka and H. Furuta, Eur. J. Org. Chem., 2005, 3887 CrossRef CAS.
- (a) T. Ishizuka, A. Osuka and H. Furuta, Angew. Chem., Int. Ed., 2004, 43, 5077 CrossRef CAS; (b) M. Toganoh, J. Konagawa and H. Furuta, Inorg. Chem., 2006, 45, 3582 CrossRef CAS.
- Direct nitration of N-confused porphyrin at the inner carbon atom has been reported: Y. Ishikawa, I. Yoshida, K. Akaiwa, E. Koguchi, T. Sasaki and H. Furuta, Chem. Lett., 1997, 26, 453 Search PubMed.
- Etheno-bridged normal porphyrins and porphycene have been synthesized previously: (a) J. Setsune, M. Ikeda, Y. Kishimoto, Y. Ishimaru, K. Fukuhara and T. Kitao, Organometallics, 1991, 10, 1099 CrossRef CAS; (b) J. Setsune and K. Hazama, Tetrahedron Lett., 1997, 38, 2513 CrossRef CAS; (c) Y. Takao, T. Takeda, K. Miyashita and J. Setsune, Bull. Chem. Soc. Jpn., 1998, 71, 1327 CrossRef CAS.
- (a) H. Furuta, T. Ishizuka, A. Osuka and T. Ogawa, J. Am. Chem. Soc., 1999, 121, 2945 CrossRef CAS; (b) H. Furuta, T. Ishizuka, A. Osuka and T. Ogawa, J. Am. Chem. Soc., 2000, 122, 5748 CrossRef CAS.
- (a) M. Toganoh, T. Ishizuka and H. Furuta, Chem. Commun., 2004, 2464 RSC; (b) M. Toganoh, S. Ikeda and H. Furuta, Chem. Commun., 2005, 4589 RSC.
- Crystal data: 1b, Violet plate, C48H34N4O, Mr = 682.79, triclinic, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 9.648(3), b = 13.477(4), c = 15.109(4) Å, α = 82.536(6), β = 74.261(6), γ = 82.981(6)°, V = 1867.2(9) Å3, Z = 2, T = 223(2) K, R = 0.0866 (I > 2σ(I)), Rw = 0.2415 (all data), GOF = 0.929. CCDC 636924. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b713845h.
(no. 2), a = 9.648(3), b = 13.477(4), c = 15.109(4) Å, α = 82.536(6), β = 74.261(6), γ = 82.981(6)°, V = 1867.2(9) Å3, Z = 2, T = 223(2) K, R = 0.0866 (I > 2σ(I)), Rw = 0.2415 (all data), GOF = 0.929. CCDC 636924. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b713845h. - Synthesis and isomerization of the imino-fused N-confused porphyrins were reported previously: N. Kashiwagi, T. Akeda, T. Morimoto, T. Ishizuka and H. Furuta, Org. Lett., 2007, 9, 1733 Search PubMed.
- Such large red-shift is not observed in the case of the etheno-bridged normal porphyrins. See ref. 9.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectroscopic data of 1, 5, 7, 8 and 9. The Cartesian coordinates of 1a, 2a and 11. See DOI: 10.1039/b713845h |
| This journal is © The Royal Society of Chemistry 2008 |