Rh-catalyzed P–P bond activation†
Stephen J.
Geier
and
Douglas W.
Stephan
*
Department of Chemistry and Biochemistry, University of Windsor, Windsor, Ontario, Canada N9B 3P4. E-mail: Stephan@uwindsor.ca; Fax: +1 519-973-7098
First published on 16th October 2007
Abstract
A Rh-catalyst derived from (NacNac)Rh(COE)(N2) effects the hydrogenation and silylation of P–P bonds to give secondary phosphines and silylphosphines, (Ph2PH) and (Ph2PSiRR′2) respectively; the latter process is shown to also involve the silylation of secondary phosphines .
The impact of organometallic chemistry and catalysis on organic chemistry has been dramatic, as is evidenced by the awarding of Nobel Prizes in 2001 and 2005. A related area, ripe for impact, involves the application of the concepts of organometallic chemistry to main group synthesis and materials chemistry. Such “inorganometallics” has drawn some recent attention.1 While much of the work to date has involved stoichiometric transformations, some catalytic processes are emerging.2–5 For example, we previously reported the use of [Cp*2ZrH3]–6–8 and CpTi(NPt-Bu3)(C2H4)2 as precatalysts for the dehydrocoupling of primary and secondary phosphines , affording a variety of P–P bonded oligomers.9 In a similar fashion, Harrod and co-workers showed that titanocene derivatives can act as catalysts for the heterocoupling of silanes and phosphines .10,11 A recent report by Waterman and co-workers highlights the catalytic heterocoupling of primary phosphines and silanes by a Zr(IV) triamidoamine complex.15 Waterman and Tilley have also reported similar use of zirconocene and hafnocene complexes to effect catalytic dehydrocoupling of stibines , affording oligostibines.12 Brookhart and Bohm have also probed similar dehydrocoupling of secondary phosphines using the Rh precatalyst Cp*Rh(CH2
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHSiMe3)2,13 although this system gave only high yields for select phosphines under harsh conditions. Most recently, Han and Tilley have reported dehydrocoupling of phosphines using a catalyst based on the Rh(iPr2PCH2CH2PiPr2) fragment.14 In related chemistry, stoichiometric functionalizations of P–P bonds have been investigated to some extent;16 metal-mediated reactions involving the activation of P–P bonds has drawn less attention. We have recently examined the stoichiometric reactivity of Ni and Fe β-diketiminates with P–H and P–P bonds.17 In this paper, we report the discovery of a catalyst for the activation of P–P bonds derived from the β-diketiminate-complex Rh(NacNac)(C8H14)N21.18
CHSiMe3)2,13 although this system gave only high yields for select phosphines under harsh conditions. Most recently, Han and Tilley have reported dehydrocoupling of phosphines using a catalyst based on the Rh(iPr2PCH2CH2PiPr2) fragment.14 In related chemistry, stoichiometric functionalizations of P–P bonds have been investigated to some extent;16 metal-mediated reactions involving the activation of P–P bonds has drawn less attention. We have recently examined the stoichiometric reactivity of Ni and Fe β-diketiminates with P–H and P–P bonds.17 In this paper, we report the discovery of a catalyst for the activation of P–P bonds derived from the β-diketiminate-complex Rh(NacNac)(C8H14)N21.18
In initial trials, we attempted to probe the utility of 1 in the dehydrocoupling of Ph2PH. Over a 12 h period at 70 °C compound 1 proved to be a poor catalyst for such dehydrocoupling, giving P2Ph4 in only 30% yield. We hypothesized that the steric demands of the NacNac ancillary ligand (NacNac = HC(CMeN(iPr2C6H2))2) favor non-productive P–H elimination over dehydrocoupling to give P–P bond formation. This supposition prompted us to probe the catalytic activity of 1 in reactions with diphosphine substrates. Treatment of P2Ph4 under 4 atm of H2 with 10 mol% of 1 at 50 °C resulted in the hydrogenation of the P–P bond to give Ph2PH in 95% yield in 12 h (Scheme 1). In a similar fashion, the product Ph2P(SiPh2H) was also prepared in 98% yield using 10 mol% 1 to catalyze the activation of P2Ph4 in the presence of 5 equivalents of Ph2SiH2 at 100 °C for 48 h (Scheme 1). Analogous use of PhMe2SiH gave the phosphine Ph2P(SiPhMe2) in 95% yield with 5% of the by-product Ph2PH, while bulkier silanes resulted in lower yields of the silylphosphine. For instance, use of Ph2MeSiH or Ph3SiH gave 76% and 74% of the P–Si coupled products Ph2P(SiPh2Me) and Ph2P(SiPh3) together with 17% and 25% yield of Ph2PH, respectively. Alkylsilanes proved to be less reactive. Et3SiH resulted in conversion of P2Ph4 to 16% of Ph2PSiEt3 with 44% Ph2PH, while iPr3SiH afforded no P–Si coupling product and only 29% Ph2PH.
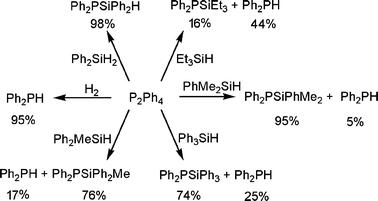 | ||
| Scheme 1 Hydrogenation and silylation of P2Ph4 catalyzed by 1. | ||
To garner further information regarding these P–P bond activations, the catalyst precursor 1 was reacted stoichiometrically with P2Ph4 in toluene and allowed to stir overnight at 25 °C. Subsequent work-up gave a dark-red residue which was recrystallized from pentane to give dark-red crystals of 2 in 50% yield.‡ It should be noted that this diminished yield is due to loss during crystallization as NMR data for reaction mixtures show near quantitative formation of 2. This product exhibits a single 31P{1H} resonance at –51.4 ppm with a Rh–P coupling of 140 Hz. The 1H NMR data for 2 was consistent with the presence of a 1 : 1 ratio of NacNac and P2Ph4. An X-ray diffraction study confirmed the formulation of 2 as Rh(NacNac)(P2Ph4) (Scheme 2, Fig. 1). The N2P2 coordination sphere about Rh is a distorted square-plane. The Rh–N distances of 2.054(3) Å and 2.065(3) Å are slightly shorter than those seen in the precursor 1 (2.076(3) Å and 2.074(3) Å) suggesting that the (P2Ph4) fragment has weaker trans influence than cis-cyclooctene and N2. The NacNac ligand bite angle in 2 is 90.53(11)°, while Rh–P distances are 2.2301(10) Å and 2.2423(10) Å. The P–P bond in 2 is 2.1389(14) Å and is comparable to the P–P bond in [Ni(NacNac)2(P2Ph2)] (2.125(3) Å).17 The RhP2 ring gives rise to a P–Rh–P angle of 57.14(4)° and thus the square-planar geometry about Rh is distorted with pseudo-trans N–Rh–P angles of 160.02(8)° and 162.37(9)° and pseudo-cis N–Rh–P angles of 107.38(9)° and 106.23(8)°. The steric crowding of the Rh coordination sphere is further evidenced from the twisting of the RhP2 plane with respect to the RhN2 plane by 14.8°. The presence of the η2-P2Ph4 to a single metal center in 2 appears to be unique in that literature precedent demonstrates the propensity of P2Ph4 to bind in a monodentate fashion, or to bridge two metal centers.19–29 In addition, the related complex Ni(NacNac)(Ph2PH)17 binds only one phosphine ligand yielding the three coordinate species, presumably a result of the slightly larger atomic radius of Rh as well as the aforementioned geometry distortions which accommodate the η2-P2Ph4.
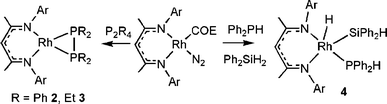 | ||
| Scheme 2 Synthesis of 2, 3 and 4. | ||
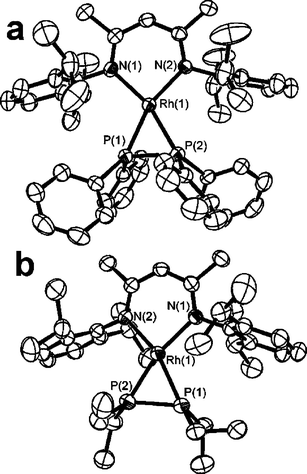 | ||
| Fig. 1 ORTEP depictions of (a) 2, and (b) 3. | ||
Reactions with P2Et4 seem to support partial dissociation of the P2 fragment as a key step in the reaction. While attempts to carry out catalytic P–P activation reactions with P2Et4 proved unsuccessful, the species Rh(NacNac)(P2Et4) 3, the analog of 2, was readily formed in a stoichiometric reaction of P2Et4 with 1 (Scheme 2). The metric parameters in 3 were found to be similar to those in 2, with Rh–N distances of 2.064(2) Å and 2.067(2) Å and Rh–P bond lengths of 2.2229(11) Å and 2.2466(10) Å, while the P–P distance was determined to be 2.1254(14) Å. These latter observations suggest that the smaller, more basic diphosphine binds to Rh more strongly, precluding the partial dissociation that initiates subsequent P–P cleavage reactions.
Addition of H2 to solutions of 2 resulted in no observable reactions at ambient temperatures, further suggesting that reaction of P2Ph4 requires thermal dissociation of the η2-P2Ph4 to at least an η1-P2Ph4 to permit oxidative addition at Rh.
In the case of H2 reactions, the transient Rh(NacNac)(η1-P2Ph4) has a vacant coordination site permitting oxidative addition of H2 to Rh. Subsequent reductive elimination of the secondary phosphine via P–P bond cleavage is proposed. Elimination of a second equivalent of phosphine regenerates the Rh(NacNac) fragment which then re-enters the catalytic cycle by reaction with P2Ph4.
In the case of the silylation reactions, an analogous catalytic cycle would generate equal amounts of Ph2PH and Ph2PSiR3 for each P2Ph4 molecule activated. However, only low concentrations of Ph2PH are observed by 31P{1H} NMR spectroscopy of the catalytic silylation reaction mixtures. This suggests that the second part of the catalytic cycle, where Ph2PH must dehydrocouple with a second equivalent of silane , is more rapid than the initial P2Ph4 activation. Reaction of P2Ph4 and two equivalents of Ph2SiH2 in the presence of 10 mol% of 1 at 50 °C for 5 days affords a 74% yield of the silylphosphine Ph2P(SiPh2H) and a 23% yield of Ph2PH.
Independent experiments demonstrated such activation of P–H bonds. For example, exposure of Ph2PH to 4 atm of D2 in the presence of 5 mol% 1 led to hydrogen for deuterium exchange resulting in 85% conversion to Ph2PD. While treatment of 1 with excess Ph2PH generates a species in solution formulated as Rh(NacNac)(Ph2PH)2, dissociation of phosphine and oxidative addition of D2, followed by reductive elimination of HD and Ph2PD accounts for the observed deuteration. Similarly, heteronuclear dehydrocoupling of Ph2PH and silanes proceeds using 5 mol% of species 1. For example, reaction of Ph2PH and Ph2SiH2 gave quantitative yield of Ph2P(SiPh2H) in 18 h at 50 °C. Similarly, Ph2P(SiPhMe2) is formed in 84% yield under similar conditions, while Ph2P(SiPh2Me) is formed in 85% yield in 18 h at 100 °C. As with the diphosphines, bulkier silanes afford lesser yields as Ph2P(SiPh3) is formed in only 40% yield from Ph2PH and Ph3SiH and the homo-dehydrocoupling by-product P2Ph4 is observed in 10% yield.
Overall, these data support a mechanism in which hydrosilylation of P2Ph4 affords both Ph2PH and the silylphosphine; however the phosphine reacts further to form another equivalent of silylphosphine (Scheme 3). This latter P–Si dehydrocoupling is apparently faster than the initial P–P bond activation. Evidence for the nature of an intermediate was derived from stoichiometric reactions of 1 with Ph2PH and Ph2SiH2. In this case a short-lived species was observed spectroscopically. The 1H NMR resonance at –13.5 ppm was indicative of a Rh–hydride species, while the doublet at 5.05 ppm indicated the presence of the PH bond of coordinated phosphine. These data together with the 31P{1H} resonance at 47.4 ppm and the 29Si{1H} signal at 21.4 ppm were consistent with the formulation of 4 as (NacNac)RhH(SiHPh2)(PHPh2) (Scheme 2). This Rh(III) intermediate is analogous to the Ir complexes (NacNac)IrH2(PR3) isolated previously by Chirik and co-workers.30 This intermediate is also related to CpRhH(SiR3)(PR3) complexes reported by Marder and co-workers.31 Loss of H2 from this species would yield the proposed intermediate (NacNac)Rh(SiHPh2)(PPh2) which is proposed to undergo reductive elimination of the silyl–phosphide product. This proposition suggests that reductive Si–P elimination occurs more readily than ligand redistribution reactions.
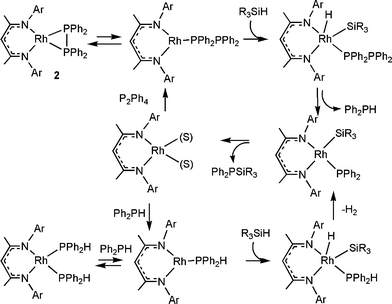
In summary, the Rh-catalyst derived from 1 effects the catalytic hydrogenation and silylation of diphosphines. Inherent in this chemistry is the dehydrocoupling of secondary phosphines with silanes . Further studies of the mechanism, catalyst optimization and applications of these processes are ongoing.
Financial support from NSERC of Canada is gratefully acknowledged. SJG is grateful for the award of an Ontario Graduate Scholarship.
Notes and references
- T. P. Fehlner, Inorganometallics, Plenum Press, New York, 1992 Search PubMed.
- F. Gauvin, J. F. Harrod and H. G. Woo, Adv. Organomet. Chem., 1998, 42, 363 CAS.
- J. Y. Corey, Adv. Organomet. Chem., 2004, 51, 1.
- T. D. Tilley, Comments Inorg. Chem., 1990, 10, 37 CAS.
- T. D. Tilley, Acc. Chem. Res., 1993, 26, 22 CrossRef CAS.
- N. Etkin, M. C. Fermin and D. W. Stephan, J. Am. Chem. Soc., 1997, 119, 2954 CrossRef CAS.
- N. Etkin, A. J. Hoskin and D. W. Stephan, J. Am. Chem. Soc., 1997, 119, 11420 CrossRef CAS.
- M. C. Fermin and D. W. Stephan, J. Am. Chem. Soc., 1995, 117, 12645 CrossRef CAS.
- J. D. Masuda, A. J. Hoskin, T. W. Graham, C. Beddie, M. C. Fermin, N. Etkin and D. W. Stephan, Chem.–Eur. J., 2006, 12, 8696 CrossRef CAS.
- R. Shu, L. Hao, J. F. Harrod, H.-G. Woo and E. Samuel, J. Am. Chem. Soc., 1998, 120, 12988 CrossRef CAS.
- S. Xin, H. G. Woo, J. F. Harrod, E. Samuel and A.-M. Lebuis, J. Am. Chem. Soc., 1997, 119, 5307 CrossRef CAS.
- R. Waterman and T. D. Tilley, Angew. Chem., Int. Ed., 2006, 45, 2926 CrossRef CAS.
- V. P. W. Bohm and M. Brookhart, Angew. Chem., Int. Ed., 2001, 40, 4694 CrossRef CAS.
- L. Han and T. D. Tilley, J. Am. Chem. Soc., 2006, 128, 13698 CrossRef CAS.
- A. J. Roering, S. N. MacMillan, J. M. Tanski and R. Waterman, Inorg. Chem., 2007, 46, 6855 CrossRef CAS.
- J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 7052 CrossRef CAS , and references therein.
- G. Bai, P. Wei, A. Das and D. W. Stephan, Dalton Trans., 2006, 1141 RSC.
- J. D. Masuda and D. W. Stephan, Can. J. Chem., 2005, 83, 324 CrossRef CAS.
- L. J. Arnold, L. Main and B. K. Nicholson, Appl. Organomet. Chem., 1990, 4, 503 CrossRef CAS.
- M. R. Churchill, C. Bueno and D. A. Young, J. Organomet. Chem., 1981, 213, 139 CrossRef CAS.
- D. Rehder, J. Organomet. Chem., 1977, 137, C25 CrossRef CAS.
- K. Issleib and G. Schwager, Z. Anorg. Allg. Chem., 1961, 310, 43 CrossRef CAS.
- A. J. M. Caffyn and M. J. Mays, J. Organomet. Chem., 2005, 690, 2209 CrossRef CAS.
- H. Adams, A. Biebricher, S. R. Gay, T. Hamilton, P. E. McHugh, M. J. Morris and M. J. Mays, J. Chem. Soc., Dalton Trans., 2000, 2983 RSC.
- A. Martin, M. J. Mays, P. R. Raithby and G. A. Solan, J. Chem. Soc., Dalton Trans., 1993, 1431 RSC.
- A. J. M. Caffyn, M. J. Mays, G. A. Solan, D. Braga, P. Sabatino, G. Conole, M. McPartlin and H. R. Powell, J. Chem. Soc., Dalton Trans., 1991, 3103 RSC.
- G. Conole, M. McPartlin, M. J. Mays and M. J. Morris, J. Chem. Soc., Dalton Trans., 1990, 2359 RSC.
- J. D. Korp, I. Bernal, J. L. Atwood, W. E. Hunter, F. Calderazzo and D. Vitali, J. Chem. Soc., Chem. Commun., 1979, 576 RSC.
- G. Fachinetti, C. Floriani and H. Stoeckli-Evans, J. Chem. Soc., Dalton Trans., 1977, 2297 RSC.
- W. H. Bernskoetter, E. Lobkovsky and P. J. Chirik, Organometallics, 2005, 24, 4367 CrossRef CAS.
- M. P. Campian, J. L. Harris, N. Jacin, R. N. Perutz, T. B. Marder and A. C. Whitwood, Organometallics, 2006, 25, 5093 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and spectroscopic details. See DOI: 10.1039/b712972f |
‡ Preparation of 2 and 3: these compounds were prepared in a similar fashion and thus only one preparation is detailed. To a solution of 30 mg (0.081 mmol) P2Ph4 in 5 mL of toluene was added a solution of 50 mg 1 (0.079 mmol) in 5 mL of toluene. The mixture was allowed to stir overnight upon which the solvent was removed in vacuo. The dark-red residue was washed with 10 mL cold pentane to give 35 mg of the product 2 (0.039 mmol, 50% yield). 1H NMR (C6D6) δ: 0.84 (6H, d, J = 6.5 Hz), 1.20 (6H, d, J = 6.5 Hz), 1.71 (6H, s), 4.45 (4H, sept, J = 6.5 Hz), 5.15 (1H, s), 6.73–7.19 (26H, br m). 31P{1H} NMR (C6D6) δ: –51.4 (d, JP–Rh = 140 Hz). 13C NMR (C6D6) δ: 24.0, 24.4, 28.6, 98.0, 124.0, 124.1, 127.5–128.6 (m, obscured by C6D6), 128.9, 134.9 (app. t, J = 7.5 Hz), 157.7, 159.6. EA anal. calcd for RhP2N2C53H61 (%) C: 71.21, H: 7.22, N: 3.13; found: C: 70.91, H: 7.22, N: 2.72. X-Ray quality crystals were grown by slow evaporation from a pentane solution. M = 890.89, space group: monoclinic, P21/n, a = 10.8591(11), b = 35.296(4), c = 12.6312(13) Å, β = 94.769(2)°, V = 4824.6(9) Å3, Z = 4, T = 273(2) K, data: variables 8472: 525, R = 0.0484, Rw = 0.1099, GOF = 1.053; CCDC 658230. 3: 1H NMR (C6D6) δ: 0.74 (8H, app. pent, J = 7.8 Hz), 1.26 (12H, t of d, J = 7.3 Hz, 2.9 Hz), 1.35 (12H, d, J = 6.9 Hz), 1.63 (12H, d, J = 6.9 Hz), 1.84 (6H, s), 4.19 (4H, sept, J = 6.9 Hz), 5.19 (1H, s), 7.15–7.29 (6H, m). 31P NMR (C6D6) δ: –64.51 (d, J = 127 Hz). 13C NMR (C6D6) δ: 9.8, 12.9, 22.4, 23.9 (d, J = 9.6 Hz), 28.3, 97.0, 123.2, 123.8, 127.3–130.5 (m, obscured by C6D6), 140.3, 156.7, 159.4. EA anal. calcd for RhP2N2C37H55 (%) C: 63.32, H: 9.19, N: 3.99; found: C: 63.55, H: 9.24, N: 4.12. X-Ray quality crystals were grown from a pentane solution at –30 °C. M = 698.73, space group: monoclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.566(3), b = 11.676(4), c = 17.636(6) Å, α = 102.412(4), β = 93.045(4), γ = 114.125(4)°, V = 1914.7(11) Å3, Z = 2, T = 296(2) K, data: variables 6726: 381, R = 0.0459, Rw = 0.1233, GOF = 0.969; CCDC 658231. For crystallographic data in CIF or other electronic format, see DOI: 10.1039/b712972f , a = 10.566(3), b = 11.676(4), c = 17.636(6) Å, α = 102.412(4), β = 93.045(4), γ = 114.125(4)°, V = 1914.7(11) Å3, Z = 2, T = 296(2) K, data: variables 6726: 381, R = 0.0459, Rw = 0.1233, GOF = 0.969; CCDC 658231. For crystallographic data in CIF or other electronic format, see DOI: 10.1039/b712972f Generation of 4: to 20 mg 1 (0.032 mmol) in 2 mL toluene was added 12 mg Ph2SiH2 (0.064 mmol). The solution was cooled to –30 °C upon which 6 mg Ph2PH (0.032 mmol) was added. The solution was allowed to warm to room temperature, volatiles were removed in vacuo and the residue was washed with pentane (2 × 2 mL), leaving 12 mg of 4 (0.013 mmol, 43% yield). 1H NMR (C6D6) δ: –13.5 (1H, d of d, JH–P = 28.4 Hz, JH–Rh = 15.4 Hz), 0.23 (3H, d, J = 6.6 Hz), 0.62 (3H, d, J = 6.6 Hz), 0.71 (3H, d, J = 6.6 Hz), 1.04 (3H, d, J = 6.6 Hz), 1.06 (3H, d, J = 6.6 Hz), 1.11 (3H, d, J = 6.6 Hz), 1.43 (3H, d, J = 6.6 Hz), 1.47 (3H, d, J = 6.6 Hz), 1.65 (3H, s), 1.88 (3H, s), 2.76 (1H, sept, J = 6.6 Hz), 2.82 (1H, sept, J = 6.6 Hz), 4.07–4.16 (2H, ov sept, J = 6.6 Hz), 5.00 (1H, d, JH–Rh = 36 Hz), 5.05 (1H, d, JP–H = 361 Hz), 5.34 (1H, s), 6.35–7.61 (26H, ov m). 31P{1H} NMR (C6D6) δ: 47.4 (d of d, JP–H = 361 Hz, JP–Rh = 137 Hz). 29Si{1H} NMR (C6D6) δ: 21.4 (d of d, J = 23.5 Hz, J = 34.1 Hz). |
| This journal is © The Royal Society of Chemistry 2008 |