Real-time monitoring of powder mixing in a convective blender using non-invasive reflectance NIR spectrometry
Luke J.
Bellamy
,
Alison
Nordon
and
David
Littlejohn
*
WestCHEM, Department of Pure and Applied Chemistry and CPACT, University of Strathclyde, 295 Cathedral Street, Glasgow, UK G1 1XL. E-mail: d.littlejohn@strath.ac.uk; Fax: +44 (0)141 548 4212
First published on 18th October 2007
Abstract
A convective blender based on a scaled down version of a high shear mixer-granulator was used to produce binary mixtures of microcrystalline cellulose (Avicel) and aspirin, citric acid, aspartame or povidone. Spectra of stationary Avicel or aspirin powder provided an indication of the information depth achieved with the NIR spectrometer used in the study, and confirmed previously reported effects of particle size and wavenumber. However, it was demonstrated that for 10% w/w aspirin in Avicel, the information depth at the C–H second overtone of aspirin (about 2.4 mm) was unaffected by changes in the particle size of aspirin and was determined by the major component. By making non-invasive NIR measurements as powders were mixed, it was possible to illustrate differences in the mixing characteristics of aspirin, citric acid, aspartame or povidone with Avicel, which were related to differences in the cohesive properties of the particles. Mixing profiles based on second overtone signals were better for quantitative analysis than those derived from first overtone measurements. It was also demonstrated that the peak-to-peak noise of the mixing profile obtained from the second overtone of aspirin changed linearly with the particle size of aspirin added to Avicel. Hence, measurement of the mixing profile in real time with NIR spectrometry provided simultaneously the opportunity to study the dynamics of powder mixing, make quantitative measurements and monitor possible changes in particle size during blending.
Introduction
Reflectance near-infrared spectrometry (NIRS) has been shown to be a useful technique for measurement of blend homogeneity during the mixing of pharmaceutical powders.1–17 NIR spectra can be measured non-invasively through a window5–7,9,13,15 or by insertion of a probe into the wall of the vessel, with the probe protruding a short distance into the powders.1–4,8 Different methods of indicating the degree of homogeneity have been reported: measurement of the change in absorbance of one of the components;9 calculation of the standard deviation2,3,6,10 or relative standard deviation8,11,12,15 for a series of consecutive measurements; calculation of the mean square of differences between consecutive spectra;17 calculation of the Euclidean distance to assess the similarity of a spectrum acquired at various points throughout the blending process with that of an ideal mixture;3,10 soft independent modelling of class analogy;3,13 bootstrap techniques;13,16 and statistical process control charts based on Hotelling's T2 statistic10 and a net analyte signal.14 Quantitative analysis of powder mixtures in blenders has been achieved by use of pre-processing methods to account for variations in light scattering and the construction of univariate6 or multivariate4,15,18 calibration models.In reflectance NIRS, the volume or mass of the powder sampled by the NIR radiation is an important consideration.19–21 There is no definitive method to determine the mass sampled by a probe, although several different procedures have been proposed.19,20 It has been suggested that no more than three times the mass of a single dosage form should be sampled.19 The information depth achieved in reflectance NIRS has an important influence on the mass of powder sampled in situ. The information depth is the distance into a sample that the NIR radiation can penetrate and still be reflected back to the spectrometer.20 A method relating the mass sampled by a fibre optic probe to spectral variance at different wavelengths was described by Cho et al.19 An increase in the estimated mass was observed as the wavelength used in the model increased, and ranged from 0.154 to 0.858 g. Berntsson et al.20 made NIR measurements of controlled depths of powders covering a strongly absorbing back plate to determine the information depth. The effects of wavelength and particle size were assessed for microcrystalline cellulose: the effective sample weight per unit area was found to be wavelength dependent with higher values obtained below 1350 nm owing to strong O–H absorbance; increasing the size of the analyte particles resulted in higher overall absorbance and greater sampled masses, although the effects of scattering were not mentioned in the discussion. Clarke et al.21 investigated the information depth by placing cellulose paper over samples of an active pharmaceutical compound; the values obtained were in the range 109–777 µm and decreased at longer wavelengths because of scattering of the incident radiation. Reflectance NIR measurements have also been obtained for tablets ranging in thickness from about 0.3 to 6.4 mm to determine the effective sample mass,22 which was dependent on the wavelength and the concentration of the active compound.
In many of these previous studies, the in-process reflectance NIR measurements were made intermittently on static powders, e.g. in between successive rotations of a tumble blender1–3,10 or the blender was stopped to allow measurements at one or more locations in the blender.5,7,13–15,17 Continuous measurements can be made during mixing if there is a suitable window6,9 or port4,8 in the vessel. However, the movement of the particles influences the NIR spectra measured and the extent of the effect depends on the timescale of the spectral scan compared to the rate of movement of the particles.23,24 With a grating spectrometer, the scan time is often too long to allow NIR measurements to be used to study particle movement.23 The shorter measurement times associated with FT-NIR spectrometry offer the opportunity to monitor particle movement and study the effect of parameters that can influence how particles mix. It has been shown that for powder velocities up to 0.73 m s–1, artefacts that are created in the interferograms from particle movement are outside the 4000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm–1 spectral region.24
000 cm–1 spectral region.24
In this study, a scaled down convective blender was used to produce binary mixtures of microcrystalline cellulose and a second compound of pharmaceutical interest. The vessel was made of glass, so it was possible to make continuous non-invasive reflectance NIR measurements of the mixing process. As a short scan time was possible with the diode array detector used, the dynamics of powder blending were monitored in real time, to ascertain how well different secondary compounds mixed with the microcrystalline cellulose. The generation of a mixing profile based on NIR spectra not only allowed quantitative measurements to be achieved, but also illustrated the influence that particle size has on the variability of the NIR mixing profile as the particles were agitated, which has not been reported in previous studies of powder blending.
Experimental
Bench top convective mixer
The design of the mixer was based on a Fielder Pharmamatrix PMA10 high shear mixer-granulator (Aeromatic-Fielder, Eastleigh, UK) and had a pot volume of approximately 500 mL. The impeller had three blades set 120° apart with a tilt angle of 45° on each blade. The motor was an IKA Eurostar overhead stirrer (VWR International) and it was positioned under the vessel to drive the impeller through a 16 ∶ 1 gearbox, giving mixing speeds up to 125 rpm. The vessel was made of glass to allow observation of the powders during mixing and to enable non-invasive measurements by NIR spectrometry (Fig. 1). One component was placed into the vessel and the stirrer motor started; agitation of the powder was monitored for a period of time to establish a baseline signal. The second compound was added by pouring powder directly from a beaker into the centre of the vessel, without stopping the mixer; mixing was then continued for a further period of time. | ||
| Fig. 1 Schematic diagram of mixing vessel and non-invasive NIR spectrometer. | ||
Powders
The microcrystalline cellulose used was Avicel grades PH-101, PH-102, PH-105 and PH-200 (FMC, Cork, Ireland) of average particle size 50, 90, 20 and 180 µm, respectively. The Avicel particles are granular in shape.Aspirin (Sigma-Aldrich, A5376, Dorset, UK), citric acid (Sigma-Aldrich, C7129) and aspartame (GlaxoSmithKline) had, respectively, the following shape and average particle size: needle (low aspect ratio)/monoclinic and 192 µm; needle (low aspect ratio) and 289 µm; needle (large aspect ratio) and 8 µm. Povidone K30 and K90 (GlaxoSmithKline) are spherical and platelet-shaped, respectively, and according to the literature,25 90% of the particles had a size >50 and >200 µm, respectively. Aspirin and Avicel were sieved through 10 cm diameter brass pan sieves (Endecotts Ltd, UK) to get smaller particle size ranges of typically <53 (<38 for Avicel), 53–106, 106–150, 150–212, 212–250, 250–300, 300–355, 355–425, 425–500, and >500 µm. The mid-point of the sieve range has been used in plots to represent each particle size fraction, except for particles <53 (<38 for Avicel) and >500 µm, where these values were used.
Near-infrared reflectance spectrometry
A Zeiss Corona 45 NIR spectrometer (Carl Zeiss, Heidenheim, Germany) was used non-invasively. The optimum measurement distance between the mixer and spectrometer was found to be 13 mm and the spot diameter was 15 mm. Measurements were acquired, relative to a reflectance standard, using Aspect software (Carl Zeiss) and stored as log1/R′. The reflectance standard for all mixing experiments was reflective white paper placed inside the mixer vessel. The integration time for each scan was set automatically; usually, NIR spectra were acquired every 0.5 s (ten co-added scans of 32 ms). A dark current measurement was required before each set of experiments to determine the inherent noise coming from the diode array detector. Spectra were exported as tab-delimited text files into Matlab version 6.5 (Mathworks, Natick, USA) for analysis using the PLS_Toolbox version 3.0.4 (Eigenvector Research, USA). First derivative spectra were calculated using the Savitsky–Golay26 algorithm with a filter width of five points and a second order polynomial. Use of second derivative spectra provided no advantage in this investigation.
Information depth for non-invasive NIR reflectance measurements
The change in the intensity of an acetate peak, from an acetate sheet positioned on top of powder layers of increasing thickness, was used to determine the information depth for Avicel. This approach was used successfully in previous studies20,21,27 when a constant signal20,27 or 50% reduction in a signal from a back plate21 was used to estimate the information depth. In this study, the constant signal procedure was used.The Zeiss Corona was positioned with the optical head pointing vertically upwards and a specifically designed adjustable PVC spacer, allowing fine tuning of the focal length and sample alignment, was fitted around the outside of the head. The optimum measurement distance (optical head to glass) was determined using a 2.68 mm layer of glass covered by a white ceramic tile; all reference measurements were made using the glass and tile combination.
A series of plastic plates, 0.25 mm thick and with a 30 mm diameter circular hole, were layered on top of the glass plate and filled with powder. The powder was carefully placed into the void, levelled off using a razor blade with minimal compaction of the sample, and the mass recorded. The assembly was placed on the spacer above the spectrometer, aligned with the incident radiation, an acetate sheet placed over the top layer of powder and the ceramic tile used as the final layer to reflect radiation back to the detector. Each NIR spectrum consisted of ten co-added scans with, in this case, an integration time of 35 ms.
Avicel (particle size <38, 150–212 and 300–355 µm) was filled into a series of ten layers of the plastic plates, with ten spectra acquired for each layer. This process was repeated three times to give a total of 30 replicate measurements per sample depth. The log1/R′ spectrum of the acetate sheet features a large peak at 6022 cm–1 and a smaller peak at 8858 cm–1; the corresponding first derivative spectrum exhibits peaks at 6086 and 8956 cm–1. There was no significant contribution from the Avicel spectrum at these wavenumbers in the first derivative spectrum. As the number of layers increased, the magnitude of the acetate signals decreased, with a constant signal indicating the information depth.
Aspirin (particle size <106, 250–300 and 425–500 µm) and binary mixtures of 10% w/w aspirin (particle size <53, 212–250 and 425–500 µm) in 75 g Avicel PH-200 were analysed using the same method. Aspirin absorbs at 6086 and 8956 cm–1 (first derivative spectrum), so the information depth was obtained from signals that increased in magnitude and then became constant as the number of layers was increased. The peak maximum for the second overtone aspirin signal in the first derivative spectra of the binary mixtures was at 8907 cm–1.
The repeatability of the filling process was found to be 4% by filling a 2.16 mm well depth ten times with Avicel (particle size 150–212 µm) and recording the mass. In contrast, the RSD values for repeat NIR measurements of a single specimen were typically less than 1%.
Real-time monitoring of mixing processes by non-invasive NIR spectrometry
75 g of Avicel PH-101 was placed in the mixing vessel and the impeller speed was set to 50 rpm. Additions of unsieved pure aspirin (0, 5, 10, 20, 30 or 40 g) were made after 120 s and mixing was continued for a further 780 s; NIR spectra were acquired every 0.5 s. The measurements in the initial 120 s were used to establish the baseline of the mixing profile prior to the addition of aspirin. The average signal during the last 99 s (i.e. 801–900 s) of the mixing profile was used for quantitative measurements at selected wavenumbers.In a second set of experiments, 15 g of unsieved powders exhibiting different particle shapes and cohesive properties (aspirin, citric acid, aspartame, povidone 30 and povidone 90) were added individually to 75 g Avicel PH-101 mixing at 50 rpm and NIR spectra were acquired every 0.5 s. Measurements in the initial 300 s were used to establish the baseline of the mixing profile prior to the addition of the secondary component; the total mixing time was 1200 s. The peaks in the first derivative spectra of the secondary components that were used to generate mixing profiles are shown in Table 1.
| Compound | Peaks in first derivative spectrum/cm–1 |
|---|---|
| Aspirin | 6086 and 8956 |
| Citric acid | 6814 and 8669 |
| Aspartame | 6043 and 8858 |
| Povidone 30 | 5960 and 8623 |
| Povidone 90 | 5960 and 8623 |
Effect of particle size of aspirin on the mixing profile
The effect of particle size on the first derivative log1/R′ mixing profiles of aspirin was investigated. 7.5 g of each particle size fraction of aspirin was added to 75 g of Avicel PH-101 mixing at 50 rpm. Additions were made after 120 s and the total mixing time was 900 s; NIR spectra were measured every 0.5 s.
Results and discussion
Information depth for non-invasive NIR reflectance measurements of static samples
Fig. 2 shows the change in the signal at 8956 cm–1 for different depths of aspirin, at three particle size ranges. The signals increased with depth as aspirin absorbs at 8956 cm–1. Reflectance and scattering is more efficient from small particles, which reduces penetration depths and explains why the signal level stabilises before that of the larger particles.28,29 The largest particles gave the most intense signals due to an increased pathlength within the sample,28,30 especially at the absorption peaks.31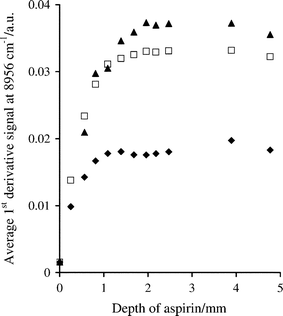 | ||
| Fig. 2 Effect of sample depth on first derivative log1/R′ spectral measurements at 8956 cm–1 for aspirin with particle size ranges of <106 (◆), 250–300 (□) and 425–500 µm (▲). | ||
Similar measurements were made for Avicel and aspirin at 6086 and 8956 cm–1 and the information depth was estimated in each case, as shown in Table 2. The information depths were different for the two compounds perhaps because of different packing characteristics of the granular and needle particles or the different densities of the powders. The results show that the information depth increases with increasing wavenumber, as noted in previous studies,20,21,27,28 which is associated with the lower sensitivity of the second overtone absorptions. Information depth values obtained from signals at 7384 cm–1 in the first derivative spectrum of Avicel were in the range 1.75–2.5 mm for the three particle sizes, intermediate between the approximate depths derived from measurements at 6086 and 8956 cm–1.
Three binary mixtures of 10% w/w aspirin (particle size <53, 212–250 and 425–500 µm) in 75 g Avicel PH-200 were prepared and subjected to the same analysis procedure as the individual compounds. The plots in Fig. 3 for the signals at 8907 cm–1 are almost identical for each particle size range of aspirin. The information depth derived from the plots in Fig. 3 is about 2.4 mm, which is comparable to that observed for Avicel sieved to a particle size range of 150–212 µm (Table 2); the average particle size of Avicel PH-200 is 180 µm. At 6086 cm–1, the information depth for each mixture was also similar, 0.8–1.1 mm, although the value was slightly less than that of Avicel at 150–212 µm, 1.3 mm. The size of the signals at 6086 cm–1 decreased as the aspirin particle size was increased, in contrast to the signals at 8907 cm–1, which showed no effect of particle size (Fig. 3).
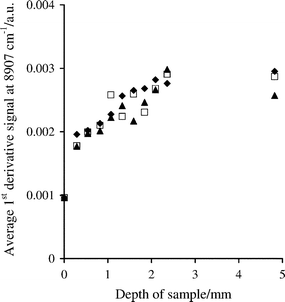 | ||
| Fig. 3 Effect of sample depth on first derivative log1/R′ spectral measurements at 8907 cm–1, for binary mixtures of 10% w/w aspirin with particle size ranges of <53 (◆), 212–250 (□) and 425–500 µm (▲) in Avicel PH-200. | ||
The experiments confirmed that the depth of sample required to achieve optical thickness increased at higher wavenumbers (e.g. for second overtone absorptions) and for larger particles of pure compounds. However, the information depth for mixtures of 10% w/w aspirin in Avicel was dependent mainly on the particle size of the main component.
Real-time monitoring of the mixing process
First derivative NIR spectra were obtained when 15 g of individual powders of different particle shape were added to 75 g Avicel PH-101 mixing at 50 rpm. Each of the added compounds generated signals at the positions of the first and second overtone absorptions listed in Table 1. When the magnitude of the signals was plotted against time, the progress of the mixing process could be monitored. Examples of the mixing profiles of aspirin, citric acid, aspartame and povidone in Avicel, based on the second overtone measurements, are shown in Fig. 4.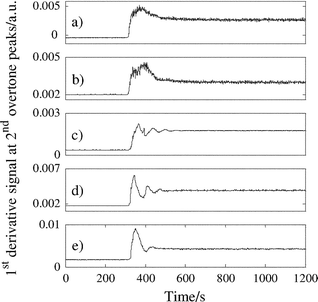 | ||
| Fig. 4 Mixing profiles for 15 g additions of (a) aspirin (8956 cm–1), (b) citric acid (8669 cm–1), (c) aspartame (8858 cm–1), (d) povidone 30 (8623 cm–1) and (e) povidone 90 (8623 cm–1) to 75 g Avicel PH-101 mixing at 50 rpm. | ||
When each of the components was added to the mixer, the powder was drawn down towards the impeller and then forced outwards and upwards against the vessel wall. This motion caused a large initial change in the measured signal as a concentrated amount of the added compound passed through the measurement zone of the spectrometer. The added component then continued to the top of the bulk powder where the mixing process continued through repeated cycles until dilution was completed, as indicated by a levelling of the NIR signal (hereafter, referred to as the plateau in the mixing profile).
Interpretation of the patterns in the mixing profiles indicates that aspirin and citric acid, which have the same particle shape, experienced a similar mixing action in the vessel. Aspartame and povidone 30 also show similar patterns in their mixing profiles; however, these materials have completely different shaped particles. Aspartame, povidone 30 and povidone 90 are not free-flowing and are prone to agglomeration: they can be termed cohesive particles; by comparison, aspirin and citric acid are both free-flowing. The mixing profiles of the more cohesive particles show more oscillations in their mixing profiles, which is probably indicative of powder dispersion occurring in stages due to poorer flow of aspartame and povidone 30 through the Avicel.
The time required for the NIR signal to become consistent did not appear to change much for the five compounds added to Avicel, which suggests that mixing in the small blender is efficient and robust to variations in particle shape and type. This may not be true at larger scale, however, where particle cohesion effects could have a more substantial influence on blending and the time taken to reach homogeneity. In this case, the improved characterisation of the mixing process provided by real-time NIR spectrometry could be beneficial.
Effect of concentration of the added component on the mixing profile
When different amounts of aspirin were added to 75 g of Avicel PH-101 mixing at 50 rpm, the mixing profiles based on the first derivative signals at 6086 and 8956 cm–1 showed similar features to those illustrated in Fig. 4, but the time required to achieve the plateau signal and the magnitude of the signal increased with the mass of the aspirin added. The plot of the average signal intensities at 8956 cm–1 (for the last 99 s of mixing) against the aspirin concentration is shown in Fig. 5.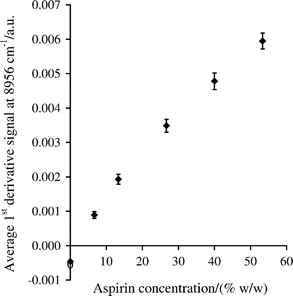 | ||
| Fig. 5 Change in average first derivative signal for aspirin in Avicel obtained from mixing profiles at 8956 cm–1, based on the last 99 s of mixing. The error bars are ± one standard deviation for n = 199. | ||
The equivalent graph for the signals at 6086 cm–1 was similar and also exhibited slight curvature, which is probably related to the relatively large information depth achieved with the Zeiss Corona and the effect of multiple absorption and scattering events as the concentration of aspirin increased. When another spectrometer and reflectance probe, which had a smaller information depth, were used to analyse the same samples, the response curves for aspirin were more linear.32
Effect of particle size of aspirin on the mixing profile
Mixing profiles were obtained when 7.5 g of nine different particle size fractions of aspirin were added to 75 g Avicel PH-101 mixing at 50 rpm. Plots of the average signal intensities (for the last 99 s of mixing) against particle size are shown in Fig. 6 for measurements made at 6086 and 8956 cm–1.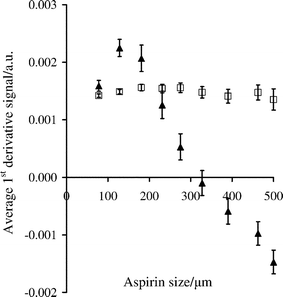 | ||
| Fig. 6 Average first derivative log1/R′ signal at 6086 (▲) and 8956 cm–1 (□) between 801 and 900 s, for mixtures of 7.5 g aspirin of varying particle size and 75 g Avicel PH-101, mixing at 50 rpm. The error bars are for ± one standard deviation based on measurements every 0.5 s giving n = 199. | ||
The average signal for the first overtone peak initially increased for aspirin particles up to 106–150 µm and then decreased as the particle size was increased further. In contrast, the plot for the second overtone peak shows very little effect of particle size on the average signal in the mixing profile. The particles under investigation are larger than the wavelength of the incident radiation, and it would be expected that the absorbance should increase with increasing aspirin particle size.30,31,33–35 As mentioned previously for static powders, the information depth of absorption measurements at the second overtone peak is larger than that of the first overtone peak. Also, the information depth in a mixture is dictated by the majority component. These observations are also likely to apply to moving particles, although the information depth will probably not be the same as that of static particles. The change in signal magnitude illustrated in Fig. 6 is likely to be related to the number of aspirin particles that the NIR radiation encounters in the sample volume, which will be greater for the smaller particle size ranges. As the first overtone peak at 6086 cm–1 has a smaller information depth, the NIR signals are more sensitive to particle size variations. The trend illustrated in Fig. 6 is consistent with the measurements at 6086 cm–1 for static mixtures of 10% w/w aspirin in Avicel, which decreased as the aspirin particle size was increased. In contrast, the larger information depth of the second overtone signals at 8956 cm–1 seems to have eliminated the effects of particle size on the measured signals, which was also observed with the static mixtures. It is probable that in the larger volume of sampled powder there is an increased probability that multiple scattering events involving the analyte particles will occur, which mitigated the effects of a reduction in the number of aspirin particles as the particle size was increased.
Although the average first derivative signal at 8956 cm–1 is relatively robust to changes in aspirin particle size, it was observed that the peak-to-peak noise of the plateau signal in the mixing profiles increased linearly with particle size, as shown in Fig. 7. This indicates that spectra were measured fast enough to note differences in short-term signal variations as aspirin particles moved into and then away from the spectrometer's observation window, with larger particles causing greater short-term variation. At 6086 cm–1, changes in the peak-to-peak noise were not as easily discerned, as the magnitude of the average signal changed with particle size of the aspirin (Fig. 6).
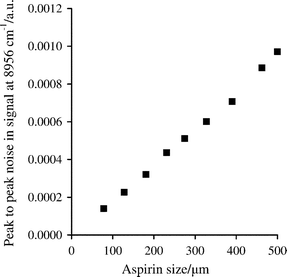 | ||
| Fig. 7 Peak-to-peak noise in first derivative NIR signals at 8956 cm–1 between 801 and 900 s, for mixtures of 7.5 g aspirin of varying particle size and 75 g Avicel PH-101 mixing at 50 rpm. | ||
Conclusions
The monitoring of powder blending in a convective mixer by non-invasive NIR spectrometry allows rapid characterisation of the mixing process and identification of the time it takes to reach homogeneity. Owing to differences in the information depth, different trends are observed for first and second overtone signals, as illustrated for additions of aspirin to Avicel. In quantitative analysis, measurements made at the second overtone peak are less sensitive to particle size changes, which make calibration functions more robust. The peak-to-peak noise of the mixing profile obtained from the second overtone of aspirin changed linearly with the particle size of aspirin, which indicates that the mixing profile can also be used to monitor particle size changes caused by particle agglomeration or attrition. Hence, measurement of the mixing profile in real time with NIR spectrometry provides simultaneously the opportunity to study the dynamics of powder mixing, make quantitative measurements of an active compound and monitor changes in particle size as blending proceeds. The methodology described has also been applied successfully to a ribbon-blender at industrial plant scale, indicating the transferability of the procedure.Acknowledgements
The support of EPSRC/DTI through LINK grant GR/R/19366/01 is acknowledged. A. N. thanks the Royal Society for a Royal Society University Research Fellowship and L. J. B. thanks CPACT for funding.References
- S. S. Sekulic, H. W. Ward, D. R. Brannegan, E. D. Stanley, C. L. Evans, S. T. Sciavolino, P. A. Hailey and P. K. Aldridge, Anal. Chem., 1996, 68, 509–513 CrossRef CAS.
- P. A. Hailey, P. Doherty, P. Tapsell, T. Oliver and P. K. Aldridge, J. Pharm. Biomed. Anal., 1996, 14, 551–559 CrossRef CAS.
- S. S. Sekulic, J. Wakeman, P. Doherty and P. A. Hailey, J. Pharm. Biomed. Anal., 1998, 17, 1285–1309 CrossRef CAS.
- O. Berntsson, L.-G. Danielsson, B. Lagerholm and S. Folestad, Powder Technol., 2002, 123, 185–193 CrossRef CAS.
- A. S. El-Hagrasy, H. R. Morris, F. D'Amico, R. A. Lodder and J. K. Drennen, J. Pharm. Sci., 2001, 90, 1298–1307 CrossRef CAS.
- D. Ely, S. Chamarthy and M. T. Carvajal, Colloids Surf., A, 2006, 288, 71–76 CrossRef CAS.
- A. S. El-Hagrasy, F. D'Amico and J. K. Drennen, J. Pharm. Sci., 2006, 95, 392–406 CrossRef CAS.
- C. Bodson, W. Dewé, Ph. Hubert and L. Delattre, J. Pharm. Biomed. Anal., 2006, 41, 783–790 CrossRef CAS.
- L. J. Bellamy, A. Nordon and D. Littlejohn, Spectrosc. Eur., 2004, 16, 30–32.
- R. De Maesschalck, F. Cuesta Sánchez, D. L. Massart and P. Doherty, Appl. Spectrosc., 1998, 52, 725–731 CrossRef CAS.
- W. Li, M. C. Johnson, R. Bruce, S. Ulrich, H. Rasmussen and G. D. Worosila, Int. J. Pharm., 2006, 326, 182–185 CrossRef CAS.
- P. E. Arratia, N. Duong, F. J. Muzzio, P. Godbole, A. Lange and S. Reynolds, Powder Technol., 2006, 161, 202–208 CrossRef CAS.
- A. S. El-Hagrasy, M. Delgado-Lopez and J. K. Drennen, J. Pharm. Sci., 2006, 95, 407–421 CrossRef CAS.
- E. T. S. Skibsted, H. F. M. Boelens, J. A. Westerhuis, D. T. Witte and A. K. Smilde, J. Pharm. Biomed. Anal., 2006, 41, 26–35 CrossRef CAS.
- A. S. El-Hagrasy and J. K. Drennen, J. Pharm. Sci., 2006, 95, 422–434 CrossRef CAS.
- D. J. Wargo and J. K. Drennen, J. Pharm. Biomed. Anal., 1996, 14, 1415–1423 CrossRef CAS.
- M. Blanco, R. Gozález Bañó and E. Bertran, Talanta, 2002, 56, 203–212 CrossRef CAS.
- O. Berntsson, L.-G. Danielsson, M. O. Johansson and S. Folestad, Anal. Chim. Acta, 2000, 419, 45–54 CrossRef CAS.
- J. Cho, P. J. Gemperline, P. K. Aldridge and S. S. Sekulic, Anal. Chim. Acta, 1997, 348, 303–310 CrossRef CAS.
- O. Berntsson, L.-G. Danielsson and S. Folestad, Anal. Chim. Acta, 1998, 364, 243–251 CrossRef CAS.
- F. C. Clarke, S. V. Hammond, R. D. Jee and A. C. Moffat, Appl. Spectrosc., 2002, 56, 1475–1483 CrossRef CAS.
- M. Iyer, H. R. Morris and J. K. Drennen, J. Near Infrared Spectrosc., 2002, 10, 233–245 Search PubMed.
- M. Andersson, O. Svensson, S. Folestad, M. Josefson and K.-G. Wahlund, Chemom. Intell. Lab. Syst., 2005, 75, 1–11 CrossRef CAS.
- O. Berntsson, L.-G. Danielsson and S. Folestad, Anal. Chim. Acta, 2001, 431, 125–131 CrossRef CAS.
- Handbook of Pharmaceutical Excipients, ed. R. C. Rowe, P. J. Sheskey and P. J. Weller, Pharmaceutical Press, London, 4th edn, 2003 Search PubMed.
- A. Savitsky and M. J. E. Golay, Anal. Chem., 1964, 36, 1627–1639 CrossRef CAS.
- O. Berntsson, T. Burger, S. Folestad, L.-G. Danielsson, J. Kuhn and J. Fricke, Anal. Chem., 1999, 71, 617–623 CrossRef CAS.
- G. Kortüm, Reflectance Spectroscopy, Principles, Methods, Applications, translated from the German by James E. Lohr, Springer-Verlag, Berlin, 1969, pp. 59–61 Search PubMed.
- C. R. Bull, J. Mod. Opt., 1990, 37, 1955–1964 CAS.
- W. W. M. Wendlandt and H. G. Hecht, Reflectance Spectroscopy, Interscience Publishers, New York, 1966, p. 51 Search PubMed.
- C. R. Bull, Analyst, 1991, 116, 781–786 RSC.
- L. J. Bellamy, Ph.D. Thesis, University of Strathclyde, UK, 2004.
- J. P. Higgins, S. M. Arrivo, G. Thurau, R. L. Green, W. Bowen, A. Lange, A. C. Templeton, D. L. Thomas and R. A. Reed, Anal. Chem., 2003, 75, 1777–1785 CrossRef CAS.
- P. Frake, I. Gill, C. N. Luscombe, D. R. Rudd, J. Waterhouse and U. A. Jayasooriya, Analyst, 1998, 123, 2043–2046 RSC.
- P. Frake, C. N. Luscombe, D. R. Rudd, J. Waterhouse and U. A. Jayasooriya, Anal. Commun., 1998, 35, 133–134 RSC.
| This journal is © The Royal Society of Chemistry 2008 |