A ‘turn-on’ FRET peptide sensor based on the mercury bindingprotein MerP
Brianna R.
White
a,
Howard M.
Liljestrand
b and
James A.
Holcombe
*a
aDepartment of Chemistry and Biochemistry, The University of Texas at Austin, Austin, TX, USA. E-mail: Holcombe@mail.utexas.edu; Fax: +1-512-471-0985; Tel: +1-512-471-5140
bDepartment of Civil, Architectural and Environmental Engineering, The University of Texas at Austin, Austin, TX, USA
First published on 12th November 2007
Abstract
A new fluorescent peptidyl chemosensor based on the mercury binding MerP protein with fluorescence resonance energy transfer (FRET) capabilities has been synthesized via Fmoc solid-phase peptide synthesis. The metal chelating unit, which is flanked by the fluorophores tryptophan (donor) and dansyl (acceptor), contains amino acids from MerP's metal binding loop (sequence: dansyl-Gly-Gly-Thr-Leu-Ala-Val-Pro-Gly-Met-Thr-Cys-Ala-Ala-Cys-Pro-Ile-Thr-Val-Lys-Lys-Gly-Gly-Trp-CONH2). A FRET enhancement or ‘turn-on’ response was observed for Hg2+ as well as for Zn2+, Cd2+ and Ag+ in a pure aqueous solution at pH 7.0. The emission intensity of the acceptor was used to monitor the concentration of these metals ions with detection limits of 280, 6, 103 and 496 µg L−1, respectively. No response was observed for the other transition, alkali and alkaline earth metals tested. The fluorescent enhancement observed is unique for Hg2+ since this metal generally quenches fluorescence . The acceptorfluorescence increase resulting from metal binding-induced FRET suggests a sensor that is inherently more sensitive than one based on quenching by the binding event.
Introduction
Determination of low level concentrations of heavy metal ions has become significant due to the severe risks they pose for human health and the environment.1 This has prompted research into the development of fluorescent chemosensors for their detection in environmental and biological samples.2–4 These chemosensors are typically composed of two covalently linked structural subunits: a fluorophore (for signal transduction) and an ionophore (for selective recognition of the metal ion). When designing these sensors, intense effort is put into maximizing the selectivity of the metal chelating unit. Chelating units composed of organic molecules have been used, but synthesis is rigorous and binding is not always reversible.3,5–15 Peptide motifs prove to be a viable alternative since they exhibit metal selectivity, can be easily synthesized via 9-fluorenyl-methoxycarbonyl (Fmoc) solid-phase peptide synthesis (SPPS)16,17 and are usable in aqueous solutions.In this study a peptide fragment based on the mercury bindingprotein MerP was used as the metal chelating unit. MerP is a member of the bacterial mercury detoxification system18,19 and is responsible for binding Hg2+ in the periplasm and transferring it to transport protein MerT. Like other metal bindingproteins , MerP contains the Cys-X-X-Cys motif and coordination to the cysteines is the dominant metal binding mechanism.20 While the full protein is 72 amino acids in length, studies have shown that the 18 residue fragment Thr-Leu-Ala-Val-Pro-Gly-Met-Thr-Cys-Ala-Ala-Cys-Pro-Ile-Thr-Val-Lys-Lys from the metal binding loop has structural and binding characteristics similar to the full protein .21,22
Typically, metal binding is detected by the quenching of a single fluorophore, e.g.dansyl,5,9,12,23–27 lucifer yellow,28anthracene.6,29–31 Although concentration-dependent quenching mechanisms have proven successful, it is inherently less sensitive than methods that producefluorescence or ‘turn-on’ as a result of binding.32 Also, it is often difficult to distinguish analyte response from sensor degradation when quenching is relied upon for quantitation.
The current study utilizes fluorescence resonance energy transfer (FRET) as a mechanism of detecting metal binding, allowing fluorescence enhancement to be monitored. Conceptually, if the chelating unit folds around the metal as it binds, the fluorophores may be brought closer together, causing increased transfer of energy from the donor fluorophore to the acceptor fluorophore. This has been successfully demonstrated by Imperiali and co-workers for Ni2+,33 and Godwin and Berg for Zn2+.34 In comparison to conventional fluorescence , FRET allows a larger wavelength separation between the excitation and emission wavelengths, thereby permitting the use of lower resolution wavelength isolation devices (e.g. filters) to measure emission without interference from the excitation source.
This paper reports on the synthesis of a new peptidyl chemosensor with FRET capabilities based on the mercury bindingprotein MerP that can be operated in aqueous solution at pH 7.0. The 23 residue peptide (sequence: dansyl-Gly-Gly-Thr-Leu-Ala-Val-Pro-Gly-Met-Thr-Cys-Ala-Ala-Cys-Pro-Ile-Thr-Val-Lys-Lys-Gly-Gly-Trp-CONH2) contains amino acids from MerP's metal binding loop. The FRET pair (tryptophan as donor and dansyl as acceptor) was conveniently attached during SPPS and are separated from the metal chelating unit by two glycine residues. Although MerP (and its metal binding loop fragment) has some binding affinity for other metals, the structures of the metal-bound forms are very different. While the protein forms a loop around the bound mercury, this loop is distorted or even non-existent with other metals.22 Because FRET is a distance-dependent interaction, the structural differences are exploited by this detection mechanism. Prior to this, there have been very few examples of Hg2+ chemosensors utilizing fluorescence enhancement due to mercury's propensity to quench fluorescence by enhanced spin–orbit coupling.35 Of these chemosensors, most are only usable in organic36–42 or mixed organic–aqueous solutions.43–50 Only four could be operated in a pure aqueous solution.51–54 Additionally, only one example of a FRET-based chemosensor for Hg2+ has been attempted, but a quenching mechanism dominated.55
Experimental
Chemicals
All chemicals were reagent grade unless noted, and deionized distilled water was used to prepare solutions. Peptide synthesis reagents N-dansyl-N′-Fmoc-ethylenediamine-MPB-AM (Dansyl NovaTag®) resin (100–200 mesh; 0.38 mmol g−1), Wang resin (100–200 mesh, 1.2 mmol g−1), glycine (Fmoc-Gly-OH), threonine [Fmoc-Thr(t-butyl ester (OtBu))-OH], leucine (Fmoc-Leu-OH), alanine (Fmoc-Ala-OH), valine (Fmoc-Val-OH), proline (Fmoc-Pro-OH), cysteine [Fmoc-Cys(Trt)-OH], isoleucine (Fmoc-Ile-OH), lysine [Fmoc-Lys(Boc)-OH], tryptophan [Fmoc-Trp(Boc)-OH] and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HBTU) were used as received from Novabiochem. All the amino acids were of L-configuration. Metal-containing solutions were prepared by dilution from 1000 µg ml−1 stock solutions. A 0.05 mol L−1 [N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)] (HEPES) (Aldrich) buffer was prepared and adjusted to pH 7.0 with ammonium hydroxide (Fisher). Other reagents used include trifluoroacetic acid (99%) (TFA) (Acros), triisopropylsilane (99%) (TIPS) (Acros), ethyl ether (Fisher), (ethylenedinitrilo)tetraacetic acid (EDTA) (EM Science), dithiothreitol (DTT) (Acros), N-methylmorpholine (NMM) (Fisher), N-methylpyrrolidone (NMP) (Fisher) and piperidine (99%) (Fisher).Apparatus
A Photon Technologies International Quanta Master Spectrofluorimeter (model QM-4/2005) was used for all fluorescence measurements.Peptide synthesis
A peptide consisting of the sequence dansyl-Gly-Gly-Thr-Leu-Ala-Val-Pro-Gly-Met-Thr-Cys-Ala-Ala-Cys-Pro-Ile-Thr-Val-Lys-Lys-Gly-Gly-Trp-CONH2 (P1) was synthesized on Dansyl NovaTag® resin and the same peptide without dansyl (P2) was synthesized on Wang resin by Fmoc solid-phase peptide synthesis using a Ranin Symphony Quartet automated peptide synthesizer. Cleavage protocols have been described earlier.56 The peptide masses (2494.3 for P1 and 2217.7 for P2) were confirmed using electrospray mass spectrometry and the purity of each sequence (63% for P1 and 73% for P2) was determined by reverse-phase HPLC . For both P1 and P2, no other single component was present in excess of 15%.Fluorescence studies
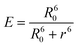 | (1) |
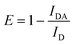 | (2) |
The distance between the fluorophores is ca. 71 Å if P1 is completely unfolded.
Results and discussion
Single-metal fluorescence response studies
For P1, the addition of Hg2+ resulted in a decrease in the tryptophan (donor) emission intensity and an increase in the dansyl emission intensity (Fig. 1a), indicating an increase in FRET due to Hg2+ binding. This response was reversible after the addition of excess EDTA. A ca. 2-fold increase (the ratio of P1 in the presence of Hg2+ to that in the absence of Hg2+) in dansyl emission was observed as well as a 35 nm blue shift in its emission to 510 nm. The blue shift in dansyl's emission is not unexpected as dansyl is an environmentally sensitive fluorophore.61 As peptide folding by metal binding occurs, the fluorophore is shielded from the polar solvent, causing a blue shift in its emission wavelength. Calibration curves were constructed from the fluorescence spectra to illustrate the quantitative dependence of the fluorescence on Hg2+ concentration. The detection limit for Hg2+ was found to be 280 µg L−1. Although this is higher than the EPA's drinking water maximum contaminant level (MCL) of 2 µg L−1,62 it is lower than41,45,50 or comparable to63 the detection limits of several existing Hg2+ fluorescent chemosensors in the literature. Additionally, this detection limit was achieved without any optimization techniques, i.e. a more intense light source, a longer integration time, etc. With optimization, this sensor could very well detect the level required by the EPA. | ||
| Fig. 1 Relationship of the fluorescence of P1 (50 mmol L−1HEPES, pH 7.0) to the concentration of a) Hg2+, b) Zn2+, c) Cd2+ and d) Ag+. λexcitation = 290 nm. All spectra were smoothed using Savitzky–Golay least squares smoothing routine with a 21-point window (Origin). | ||
Addition of Cd2+, Zn2+and Ag+ to P1 also resulted in a metal binding-induced FRET response (Fig. 1b–d) and this response was reversible after the addition of excess EDTA. The largest FRET increase of any metal was observed for Zn2+, which caused a ca. 11-fold increase in dansyl emission along with a 47 nm blue shift of the peak emission to 498 nm. The detection limit for Zn2+ was found to be 6 µg L−1, well below the EPA's drinking water MCL of 5 mg L−1.64Cd2+ addition resulted in the second largest FRET response. A ca. 6-fold increase in dansyl emission and a 45 nm blue shift to 500 nm was observed. The detection limit of Cd2+ was found to be 103 µg L−1. While this is above the EPA drinking water MCL of 5 µg L−1,64 it is only the third example of a fluorescence sensor that utilizes enhancement for the detection of Cd2+. Additionally, the focus of the previous examples was not on metal detection, but rather on using enhancement to monitor protein conformational changes65 and the synthesis of a dual receptor system for general sensing of anions and cations.66 Similarly to Hg2+, Ag+ addition resulted in a ca. 2-fold increase in dansyl emission intensity. A 39 nm blue shift to 506 nm was also observed. The detection limit for Ag+ was found to be 496 µg L−1. A summary of these results is shown in Table 1.
| Metal | λ emission/nma | Enhancement factorb | R/Åc | Detection limit/µg L−1 | Log Kd |
|---|---|---|---|---|---|
| a Dansyl emission peak after addition of metal. The P1 emission intensity for dansyl chloride in the absence of metal is 545 nm. b Ratio of the intensity of P1 in the presence of metal ion to that in the absence of metal ion. c The distance between the fluorophores (R) before the addition of metal is 21.0 ± 0.6 Å. d Metal binding constant was obtained via non-linear fitting of the fluorescence titration data. Although P1 contained more than one site for these metals, only one could be determined. | |||||
| Hg2+ | 510 | 2 | 19.4 ± 0.6 | 280 | 4.4 |
| Zn2+ | 498 | 11 | 16.6 ± 0.7 | 6 | 5.2 |
| Cd2+ | 500 | 6 | 17.9 ± 0.5 | 103 | 5.9 |
| Ag+ | 506 | 2 | 19.1 ± 0.6 | 496 | 5.4 |
Although this binding motif was taken from the mercury bindingprotein MerP, it is not surprising that it binds other metals in addition to Hg2+. The Cys-X-X-Cys motif present in MerP is also found in many other soft-metal bindingproteins including cadmium transportprotein CadA,67 the zinc finger domains68 and superoxide dismutase, a yeast copper- and zinc-transporting protein .69 Additionally, studies conducted by Opella and co-workers on MerP's metal binding loop fragment showed affinity for Zn2+ (log K = 3.5), Cd2+ (log K = 3.4) and Ag+ (log K = 3.4), which are not significantly different than that found for Hg2+ (log K = 4.0).22 It is curious that this sensor does not have a larger response to Hg2+, but this may be due to the apparent quenching of the Trpdonor by Hg2+, which will be discussed further.
The fluorescence response of P1 for a number of metal ions is presented in Table 2. P1 had no response to other transition, alkali and alkaline earth metals tested, illustrating its potential use in a variety of matrices. In addition to selectivity, the robustness of the sensor was also evaluated by varying the pH. When the pH was lowered to 3.5, dansyl's emission intensity decreased noticeably by a factor of ca. 5, but was restored when the pH was returned to 7.0.
Evaluation of conditional stability constants
Fig. 2a–d shows that the maximum metal : peptide binding ratio is approximately 3 for Hg2+ and Zn2+ and 2 for Cd2+ and Ag+. Opella and co-workers previously reported a metal : peptide binding ratio of 1 for all of these metals.22 It is possible that the additional residues (four glycines and two fluorophores) in this study could allow for more binding sites or change the structure of the peptide such that other residues are available for binding. | ||
| Fig. 2 Fluorescent binding curves of P1 with a) Hg2+, b) Zn2+, b) Cd2+ and d) Ag+. λexcitation = 290 nm. The metal to peptide binding ratio is 2 : 1 for Cd2+ and Ag+ and 3 : 1 for Hg2+ and Zn2+. | ||
While the FRET signal increases with metal concentration in all cases, it would not be surprising that the acceptor-to-donor distances would be different depending on whether one or two metals were bound. Hence the fluorescence sensitivity could very likely change whether P1 is binding one, two or three cations. Thus, it is difficult to confidently state that the proportionality constant relating the signal to the amount of metal bound has a constant value. For example, looking at the response for Zn2+ (Fig. 2b), it is relatively obvious that binding the first metal (0–10 µM) produces a smaller FRET signal than when the second metal is bound (ca. 10–20 µM). Deconvoluting the changing response is difficult but necessary if one were to attempt to develop an isotherm from which to extract log K values. In contrast to Zn2+, Cd2+ binding (Fig. 2c) appears to be relatively well behaved in spite of the obvious 2 : 1 metal : peptide ratio at saturation.
In an attempt to get some binding information, the data in Fig. 2 were used to construct binding isotherms based on a one-site model. Using Graphpad Prism 4®, conditional stability constants were calculated for each metal. Although P1 bound these metals in ratios greater than one, only one binding site log K was determined due, in part, to the shape of the binding curve. From a fit of the data, the estimated log K is 4.4 for Hg2+, 5.2 for Zn2+, 5.9 for Cd2+ and 5.4 for Ag+. Similarly to previous reports,22 the values for Zn2+, Cd2+ and Ag+ are not significantly different. While goodness-of-fit was poorest for Hg2+, an R2 value of 0.999 was obtained for Cd2+ binding.
When looking at Table 1, it is interesting to note that these log K values do not match the amount of FRET enhancement. For instance, Zn2+ binding resulted in the largest enhancement, but its log K is smaller than Cd2+ and Ag+. This is not surprising since there is no assurance that stronger binding would necessarily bring the fluorophores in closer proximity. Additional structural studies to elucidate the conformation of the peptide with and without a particular metal will be needed to make a more definitive statement.
Determination of FRET efficiency
Using eqn (1), the FRET efficiency without metal was determined to be 49 ± 4%, which corresponds to a distance of only 21.0 ± 0.6 Å between the fluorophores, indicating that P1 (71 Å when completely elongated) is coiled or folded even before the addition of the metal.Eqn (1) was also used to calculate the FRET efficiency and distance between the fluorophores for P1 after metal binding, and these values are reported in Table 1. As expected, a large change in distance was seen for Zn2+ and Cd2+ which had large FRET enhancement. Hg2+ and Ag+, which had the smallest enhancement, also had small distance changes. The small FRET efficiency increase for Hg2+ may be explained by its propensity to coordinate with amines .70,71 When monitoring P2, which contains just the Trpdonor, its emission was quenched to 30.7% of its initial value at 30 µmol L−1Hg2+. It has been shown that Hg2+ complexes with Trp's indole ring, causing quenching .72 If this occurs, the amount of energy that Trp is able to transfer to dansyl will yield an apparent reduction in the FRET signal. Additionally, Hg2+ binding to other amine -containing sites may prevent the peptide from obtaining the loop conformation described by Opella and co-workers,22 which could also result in a lower FRET efficiency. It should be noted that no quenching of dansyl by Hg2+ was observed when P1 was excited only at dansyl's absorption maximum.
Evaluation of mixed Hg2+, Zn2+ and Cd2+ solutions
In order to determine the response of P1 to mixed solutions of Hg2+, Zn2+ and Cd2+, various ratios of the metals were added to P1 solutions. Initially, the enhancement and blue shift in dansyl's emission intensity for two metal solutions were monitored. Various ratios of Cd2+ : Zn2+ produced an enhancement factor and blue shift similar to what was observed for just Cd2+, which is expected since Cd2+ had a larger log K value. Various ratios of Hg2+ : Zn2+ and Hg2+ : Cd2+ produced an enhancement factor and blue shift similar to what was observed for Hg2+. Although Zn2+ and Cd2+ both individually had better FRET responses than Hg2+, Hg2+quenching of Trp may have the largest effect on the FRET response of P1. This same result was also observed when all three metals were simultaneously monitored.Conclusions
A new fluorescent peptidyl chemosensor based on the mercury bindingprotein MerP with FRET capabilities was designed and quantitatively characterized. This study represents the first FRET-based fluorescence enhancement with the binding of Hg2+, a metal that is generally characterized by its propensity to quench fluorescence . As a general rule, the appearance of a fluorescence signal with a binding event will be inherently more sensitive than an approach based on quenching by an analyte due to shot noise. Unlike many previous examples of Hg2+ sensors, the peptidyl chemosensor functioned in aqueous solution at pH 7.0Responses to Zn2+, Cd2+and Ag+ were also observed, with Zn2+ and Cd2+ binding producing the largest FRET enhancement response. No FRET response to the other transition, alkali and alkaline earth metals tested was observed. Although one might intuitively think that the largest response should come from Hg2+, the response is based on the change in proximity of the fluorophores as a result of metal binding and not on binding strength. Additionally, Hg2+-induced quenching of the Trpdonor as well as Hg2+ binding to amine -containing sites may be limiting the amount of FRET that can occur. A larger FRET enhancement for Hg2+ could potentially be obtained by using a different FRET pair. While two UV-excitable fluorophores were used in this study, other FRET pairs usable in the visible or infrared region could feasibly be implemented for in situ applications.
Acknowledgements
This work was supported, in part, by the Robert A. Welch Foundation and the Texas Hazardous Waste Research Center. We would like to thank Professor Jason Shear for use of the spectrofluorimeter and Klaus Linse, Sandra Smith and Michelle Gadush for their assistance in peptide synthesis.References
- L. Jarup, Br. Med. Bull., 2003, 68, 167–182 Search PubMed.
- L. Fabbrizzi and A. Poggi, Chem. Soc. Rev., 1995, 24, 197–202 RSC.
- R. Kramer, Angew. Chem., Int. Ed., 1998, 37, 772–773 CrossRef CAS.
- R. E. Williams, P. J. Holt, N. C. Bruce and C. R. Lowe, in Biosensors for Environmental Monitoring, ed. U. Bilitewski and A. P. F. Turner, Harwood Academic Publishers, Amsterdam, 2000, pp. 213–225 Search PubMed.
- R. Corradini, A. Dossema, G. Galaverna, R. Marchelli, A. Panagia and G. Sartor, J. Org. Chem., 1997, 62, 6283–6289 CrossRef CAS.
- G. De Santis, L. Fabbrizzi, M. Licchelli, C. Mangano, D. Sacchi and N. Sardone, Inorg. Chim. Acta, 1997, 257, 69–76 CrossRef CAS.
- J. Yoon, N. E. Ohler, D. H. Vance, W. D. Aumiller and A. W. Czarnik, Tetrahedron Lett., 1997, 38, 3845–3848 CrossRef CAS.
- K. A. Mitchell, R. G. Brown, D. Yuan, S. C. Chang, R. E. Utecht and D. E. Lewis, J. Photochem. Photobiol., A, 1998, 115, 157–161 CrossRef CAS.
- L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, Eur. J. Inorg. Chem., 1999, 455–460 CrossRef CAS.
- M. Beltramello, M. Gatos, F. Mancin, P. Tecilla and U. Tonellato, Tetrahedron Lett., 2001, 42, 9143–9146 CrossRef CAS.
- G. Klein, D. Kaufmann, S. Schurch and J.-L. Reymond, Chem. Commun., 2001, 561–562 RSC.
- L. Prodi, M. Montalti, N. Zaccheroni, F. Dallavalle, G. Folesani, M. Lanfranchi, R. Corradini, S. Pagliari and R. Marchelli, Helv. Chim. Acta, 2001, 84, 690–706 CrossRef CAS.
- J. Bourson, F. Badaoui and B. Valeur, J. Fluoresc., 1994, 4, 275–277 CAS.
- E. U. Akkaya and S. Turkyilmaz, Tetrahedron Lett., 1997, 38, 4513–4516 CrossRef CAS.
- J. Kawakami, H. Itoh, H. Mitsuhashi and S. Ito, Anal. Sci., 1999, 15, 617–618 CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- E. Atherton and R. C. Sheppard, Solid Phase Peptide Synthesis: A Practical Approach, IRL Press, Oxford, 1989, pp. 131–148 Search PubMed.
- N. L. Brown, Trends Biochem. Sci., 1985, 10, 400–403 CrossRef CAS.
- T. J. Foster, CRC Crit. Rev. Microbiol., 1987, 15, 117–140 CrossRef CAS.
- S. J. Opella, T. M. DeSilva and G. Veglia, Curr. Opin. Chem. Biol., 2002, 6, 217–223 CrossRef CAS.
- G. Veglia, F. Porcelli, T. DeSilva, A. Prantner and S. J. Opella, J. Am. Chem. Soc., 2000, 122, 2389–2390 CrossRef CAS.
- T. M. DeSilva, G. Veglia, F. Porcelli, A. Prantner and S. J. Opella, Biopolymers, 2002, 64, 189–197 CrossRef CAS.
- A. Torrado, G. K. Walkup and B. Imperiali, J. Am. Chem. Soc., 1998, 120, 609–610 CrossRef CAS.
- S. Bhattacharya and M. Thomas, Tetrahedron Lett., 2000, 41, 10313–10317 CrossRef CAS.
- A. Singh, L. Yao, W. C. Still and D. Sames, Tetrahedron Lett., 2000, 41, 9601–9605 CrossRef CAS.
- Y. Zheng, K. M. Gattas-Asfura, V. Konka and R. M. Leblanc, Chem. Commun., 2002, 2350–2351 RSC.
- Y. Zheng, X. Cao, J. Orbulescu, V. Konka, F. M. Andreopoulos, S. M. Pham and R. M. Leblanc, Anal. Chem., 2003, 75, 1706–1712 CrossRef CAS.
- T. Mayr and T. Werner, Analyst, 2002, 127, 248–252 RSC.
- P. Jiang and Z. Guo, Coord. Chem. Rev., 2004, 248, 205–229 CrossRef CAS.
- L. Fabbrizzi, M. Licchelli, P. Pallavicini, A. Perotti, A. Taglietti and D. Sacchi, Chem.–Eur. J., 1996, 2, 75–82 CrossRef CAS.
- L. Fabbrizzi, M. Licchelli, P. Pallavicini, A. Perotti and D. Sacchi, Angew. Chem., Int. Ed. Engl., 1994, 33, 1975–1977 CrossRef.
- M. Y. Chae and A. W. Czarnik, J. Am. Chem. Soc., 1992, 114, 9704–9705 CrossRef CAS.
- D. A. Pearce, G. K. Walkup and B. Imperiali, Bioorg. Med. Chem. Lett., 1998, 8, 1963–1968 CrossRef CAS.
- H. A. Godwin and J. M. Berg, J. Am. Chem. Soc., 1996, 118, 6514–6515 CrossRef.
- D. S. McClure, J. Chem. Phys., 1952, 20, 682–686 CAS.
- G. Hennrich, H. Sonnenschein and U. Resch-Genger, J. Am. Chem. Soc., 1999, 121, 5073–5074 CrossRef CAS.
- K. Rurack, M. Kollmannsberger, U. Resch-Genger and J. Daub, J. Am. Chem. Soc., 2000, 122, 968–969 CrossRef CAS.
- K. Rurack, U. Resch-Genger, J. L. Bricks and M. Spieles, Chem. Commun., 2000, 2103–2104 RSC.
- G. Hennrich, W. Walther, U. Resch-Genger and H. Sonnenschein, Inorg. Chem., 2001, 40, 641–644 CrossRef CAS.
- J. V. Mello and N. S. Finney, J. Am. Chem. Soc., 2005, 127, 10124–10125 CrossRef CAS.
- R. Martinez, A. Espinosa, A. Tarraga and P. Molina, Org. Lett., 2005, 7, 5869–5872 CrossRef CAS.
- H. Zhang, L. Han, K. A. Zachariasse and Y. Jiang, Org. Lett., 2005, 7, 4217–4220 CrossRef CAS.
- L. Prodi, C. Bargossi, M. Montalti, N. Zaccheroni, N. Su, J. S. Bradshaw, R. M. Izatt and P. B. Savage, J. Am. Chem. Soc., 2000, 122, 6769–6770 CrossRef CAS.
- X. Guo, X. Qian and L. Jia, J. Am. Chem. Soc., 2004, 126, 2272–2273 CrossRef CAS.
- A. Caballero, R. Martinez, V. Lloveras, I. Ratera, J. Vidal-Gancedo, K. Wurst, A. Tarraga, P. Molina and J. Veciana, J. Am. Chem. Soc., 2005, 127, 15666–15667 CrossRef CAS.
- G. Zhang, D. Zhang, S. Yin, X. Yang, Z. Shuai and D. Zhu, Chem. Commun., 2005, 2161–2163 RSC.
- Y. Yang, K. Yook and J. Tae, J. Am. Chem. Soc., 2005, 127, 16760–16761 CrossRef CAS.
- Y. Zhao, Z. Lin, C. He, H. Wu and C. Duan, Inorg. Chem., 2006, 45, 10013–10015 CrossRef CAS.
- H. Zheng, Z. Qian, L. Xu, F. Yuan, L. Lan and J. Xu, Org. Lett., 2006, 8, 859–861 CrossRef CAS.
- S. H. Kim, J. S. Kim, S. M. Park and S. Chang, Org. Lett., 2006, 8, 371–374 CrossRef CAS.
- E. M. Nolan and S. J. Lippard, J. Am. Chem. Soc., 2003, 125, 14270–14271 CrossRef CAS.
- E. M. Nolan, M. E. Racine and S. J. Lippard, Inorg. Chem., 2006, 45, 2742–2749 CrossRef CAS.
- S.-K. Ko, Y.-J. Yang, J. Tae and I. Shin, J. Am. Chem. Soc., 2006, 128, 14150–14155 CrossRef CAS.
- S. Yoon, A. Albers, A. P. Wong and C. J. Chang, J. Am. Chem. Soc., 2005, 127, 16030–16031 CrossRef CAS.
- A. Ono and H. Togashi, Angew. Chem., Int. Ed., 2004, 43, 4300–4302 CrossRef CAS.
- B. R. White and J. A. Holcombe, Talanta, 2007, 71, 2015–2020 CrossRef CAS.
- T. Forster, Ann. Phys., 1948, 2, 55–75 Search PubMed.
- B. W. Van Der Meer, G. Coker, III and S. Y. S. Chen, Resonance Energy Transfer: Theory and Data, VCH Publishers, Inc., New York, 1994, p. 157 Search PubMed.
- B. M. Dunn, C. Pham, L. Raney, D. Abayasekara, W. Gillespie and A. Hsu, Biochemistry, 1981, 20, 7206–7211 CrossRef CAS.
- M. Gustiananda, J. R. Liggins, P. L. Cummins and J. E. Gready, Biophys. J., 2004, 86, 2467–2483 CrossRef CAS.
- J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, Plenum Press, New York, 1st edn, 1983, pp. 189–218 Search PubMed.
- US Environmental Protection Agency, Mercury Update: Impact on Fish Advisories, EPA Fact Sheet: EPA-823-F-01-011, EPA Office of Water, Washington, DC, 2001 (http://www.epa.gov/waterscience/fishadvice/mercupd.pdf) Search PubMed.
- A. B. Descalzo, R. Martinez-Manez, R. Radeglia, K. Rurack and J. Soto, J. Am. Chem. Soc., 2003, 125.
- US EPA, http://www.epa.gov/safewater/contaminants/index.html#mcls, 2003.
- S.-H. Hong and W. Maret, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2255–2260 CrossRef CAS.
- F. Oton, A. Tarraga and P. Molina, Org. Lett., 2006, 8, 2107–2110 CrossRef CAS.
- G. Nucifora, L. Chu, T. K. Misra and S. Silver, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 3544–3548 CrossRef CAS.
- J. M. Berg, Acc. Chem. Res., 1995, 28, 14–19 CrossRef CAS.
- V. P. Culotta, L. W. J. Klomp, J. Strain, R. L. B. Casareno, B. Krems and J. D. Gitlin, J. Biol. Chem., 1997, 272, 23469–23472 CrossRef CAS.
- J.-M. Lehn and F. Montavon, Helv. Chim. Acta, 1978, 61, 67–82 CrossRef CAS.
- N. R. Cha, M. Y. Kim, Y. H. Kim, J.-I. Choe and S.-K. Chang, J. Chem. Soc., Perkin Trans. 2, 2002, 1193–1196 RSC.
- R. F. Chen, Arch. Biochem. Biophys., 1971, 142, 552–564 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |