Direct acoustic profiling of DNA hybridisation using HSV type 1 viral sequences†
Yıldız
Uludağ
a,
Xin
Li
a,
Heather
Coleman
b,
Stacey
Efstathiou
b and
Matthew A.
Cooper
*a
aAkubio Ltd, 181 Cambridge Science Park, Cambridge, UK CB4 0GJ. E-mail: mcooper@akubio.com; Fax: +44 (0) 1223 225336; Tel: +44 (0) 1223 225326
bDepartment of Pathology, Cambridge University, Tennis Court Road, Cambridge, UK CB2 1QP
First published on 21st September 2007
Abstract
We describe the detection of specific, conserved DNA sequences of herpes simplex virus (HSV) type 1 by means of a novel, high sensitivity acoustic biosensor . Repeated assays on planar and polymeric carboxylic acid - and biotin-presenting surface chemistries enabled statistical comparison of assay specificity and sensitivity and evaluation of assay Z-factor scores. Using a three minute hybridisation with NeutrAvidin capture for signal enhancement, it was possible to detect HSV viral nucleic acids at 5.2 × 10−11 M concentration.
Introduction
Nucleic acid -based recognition of viral sequences provides a precise and accurate method for clinical screening, diagnosis and monitoring the efficacy of anti-viral therapy. Alternative methods such as cell culture detection and enzyme-linked immunosorbent assay (ELISA) are slow in comparison.1 Most nucleic acid -based tests utilise PCR to amplify nucleic acids and use fluorophore labels for detection. Real-time, label-free biosensors have the potential to provide an alternative to the current nucleic acid -based tests if the required sensitivity is reached and the size of the biosensors is miniaturised.Biosensors using quartz crystal microbalance (QCM) technology provide a label-free method for detecting and analysing molecules and, unlike more complex optical biosensors , QCM devices have the prospect of miniaturisation. Therefore QCM sensors provide an ideal platform for nucleic acid -based recognition. The first nucleic acid detection using QCM technology was performed by Fawcett et al.2 Fawcett and colleagues immobilised single-stranded DNA probes on the sensor and observed the hybridisation of the target as a frequency decrease. Since this first example, several papers have been published on nucleic acid detection using QCM devices. For example Tombelli et al. developed a QCM biosensor for a specific DNA sequence from methicillin-resistant Staphylococcus aureus (MRSA)3 – a major cause of hospital infections. They detected both synthetic nucleotides and PCR products with a concentration of 3 × 10−8 M PCR products resulting in a 38 ± 3 Hz frequency change.
In the current study, we describe the detection of specific, conserved DNA sequences of herpes simplex virus (HSV) type 1. HSV causes recurrent mucosal infections of the eye, mouth and genital tract. HSV type 1 establishes a lifelong latent infection within the host which can subsequently reactivate to cause recurrent infections and occasionally life-threatening HSV encephalitis. Following infection the virus gains access to sensory nerve terminals and latency is established in corresponding sensory neurones.4 Two different probe and complementary target sets were used for the HSV recognition assays that are from ICP0 and VP16 generegions of the HSV viral sequence. ICP0 is one of the infected-cell proteins (ICPs) that accumulate in infected cells but is absent from uninfected cells. ICP0 activates genes introduced into cells by transfection or infection.5 VP16 is an essential structural protein and also functions as a major virion transactivator of virusgene expression. VP16 interacts with two cellular proteins , Oct-1 and HCF (host cell factor), and this complex binds to TAATGARAT motifs to activate the transcription of the viral immediate-early genes.6 HSV regulatory proteins VP16 and ICP0 play key roles to stimulate viral gene expression during the earliest stages of infection;7 thus, clinically it is relevant to diagnose HSV infection by detecting the genes encoding VP16 or ICP0 since these are important replication and virulence determinants.
In an earlier study, Napier et al. used an electrochemical sensor to detect synthetic DNA and genomic DNA from HSV type 2 after PCR amplification.8 In another study, Kara et al. used an electrochemical sensor to detect and differentiate HSV type 1 and type 2 viral sequences using both synthetic and PCR amplified real samples.9 In the current study, a RAP♦id instrument was used as the biosensor platform for the detection of HSV type 1 viral sequences. One notable advantage of RAP™ (Resonant Acoustic Profiling™) detection over more established optical label-free detection is the relative insensitivity of acoustics to changes in solvent/medium when running samples in complex media such as serum , plasma or whole blood. Optical detection systems suffer from large bulk shifts which need to be minimised by calibration routines and dilution of the sample. In contrast, acoustic systems are not affected by refractive index changes, but are instead sensitive to bulk effects dominated by the viscosity and density of the media.10 Thus viral detection from clinical samples with minimal sample processing is feasible when acoustic sensors are employed for nucleic acid testing. The RAP♦id system possesses multi-channel sensors, a low-stress crystal mount, microfluidics, higher fundamental frequency sensors and automated sample handling, which together enhance system sensitivity and robustness.
The sensitivity of oligonucleotide hybridisation achieved by QCM sensors has been previously reported to be ca. 10−8 M.11–13 Nanoparticles are commonly used to enhance the sensitivity of hybridisation to the region of 10−15–10−16 M.14–16 In this paper we show that without the use of nanoparticles but utilising capture protein , a hybridisation sensitivity of 10−11 M can be achieved.
For this study, we have compared three different surface chemistries according to their hybridisation performances. The optimised surface chemistry was then combined with a simple signal amplification method to enhance the limit of detection for HSV viral DNA recognition.
Experimental
RAP experiments were conducted using an automated four-channel RAP♦id 4 instrument (RAP♦id 4; Akubio Ltd, Cambridge, UK). The instrument applies the principles of QCM, in that a high frequency (16.5 MHz) voltage is applied to a piezoelectric quartz crystal to induce the crystal to resonate, and its resonance frequency is then monitored in real time. RAP♦id 4 integrates acoustic detection with a continuous flow microfluidic delivery system, a thermal control unit, and automated sample handling. Four individual flow cells enable up to four measurements to be performed simultaneously. The volume of the flow cell used in this study was 900 nl. The exchange rate in the flow cells could be set as slow as 2 s at a flow rate of 25 µl min−1 and as fast as 14 ms at a flow rate of 4000 µl min−1. To conserve sample, 25 µl min−1 was employed for the pathfinder assay development. Baseline drift observed during the study was 0.25 ± 0.15 Hz (n = 12) after docking and priming the sensor chips. The operating temperature was 25 ± 0.5 °C throughout the assays.Sensor chip fabrication
AKT♦iv Covalent sensor chips were fabricated using a proprietary protocol and carboxymethyldextran-T500 (CMD-T500) polymer sensor chips were fabricated according to the protocol described by Li et al.17Fabrication of biotin chips was as follows, with all operations carried out in a class 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cleanroom. Gold-coated 17 MHz quartz sensor chips were cleaned using a proprietary plasma etching process in a PT7160 RF Plasma Barrel etcher (Quorum Technologies, Newhaven, UK) for 55 s. The cleaned quartz resonators were then submerged in an aqueous solution of 0.5 mM biotin-PEG-disulfide (Polypure, Oslo, Norway) overnight. After rinsing with water, the sensor chips were dried and assembled into sensor cassettes. Biotin-coated sensors were stored in the dark under nitrogen until required.
000 cleanroom. Gold-coated 17 MHz quartz sensor chips were cleaned using a proprietary plasma etching process in a PT7160 RF Plasma Barrel etcher (Quorum Technologies, Newhaven, UK) for 55 s. The cleaned quartz resonators were then submerged in an aqueous solution of 0.5 mM biotin-PEG-disulfide (Polypure, Oslo, Norway) overnight. After rinsing with water, the sensor chips were dried and assembled into sensor cassettes. Biotin-coated sensors were stored in the dark under nitrogen until required.
Sensor surface preparation for AKT♦iv Covalent sensor chips
AKT♦iv Covalent sensor chips (Akubio Ltd, Cambridge, UK) were employed for the assays. Sensor surfaces were prepared by immobilising NeutrAvidin (NA; Perbio Science UK Ltd, Cramlington, UK) on sensors using conventional amine coupling chemistry. Running buffer used for immobilisation was degassed Dulbecco's modified phosphate buffered saline (PBS, pH 7.4; Sigma-Aldrich, Poole, UK). The flow rate of the buffer for the assay was 25 µl min−1. Sensor surfaces were first activated with a 1 : 1 mixture of 400 mM 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) and 100 mM N-hydroxysuccinimide (NHS) (LINK♦it Coupling Solution kit; Akubio Ltd, Cambridge, UK), prepared in 0.22 µm-filtered deionised water, and mixed immediately prior to use (final concentrations: 200 mM EDC and 50 mM NHS). EDC–NHS was injected simultaneously across all four sensor surfaces for 3 min. NA (50 µg ml−1 in PBS buffer) was then injected simultaneously across the sensor surfaces for 3 min. Non-reacted NHSesters were capped with 1 M ethanolamine, pH 8.5 (LINK♦it Coupling Solution kit; Akubio Ltd, Cambridge, UK). The frequency changes were recorded 2 min after the protein injection was completed. After NA immobilisation, the running buffer was changed to Tris buffer comprising 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, pH 7. The biotinylated complementary surface probe and scrambled surface probe (biotinylated probes; TIB Molbiol, Berlin, Germany) were diluted in Tris buffer to 10 µg ml−1 and injected separately over different flow cells for 3 min to create active and control surfaces. The frequency changes of the biotinylated probes captured were recorded 4 min after the end of the injection.Sensor surface preparation for CMD-T500 polymer sensor chips
The protocol used for DNA capture and hybridisation on CMD-T500 polymer sensor chips was the same as for AKT♦iv Covalent sensor chips, except that immobilisation of NA was performed using 10 mM phosphate buffer pH 6.5 instead of PBS buffer pH 7.4.Sensor surface preparation for biotin sensor chips
Running buffer used for immobilisation was PBS and the flow rate was 25 µl min−1. NA was prepared for immobilisation at 50 µg ml−1 in PBS, and injected simultaneously across separate sensor surfaces of the biotin chips for 3 min. A solution of 0.1 M HCl was then injected for 1 min to remove the loosely bound excess protein from the surface. The immobilisation level was recorded 1 min after HCl injection as a frequency change. After NA capture on the biotin surface, running buffer was changed to Tris buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, pH 7). The biotinylated complementary surface probe and scrambled surface probe (biotinylated probes; TIB Molbiol, Berlin, Germany) (Table 1) were diluted in Tris buffer to 10 µg ml−1 and each injected over separate flow cells for 5 min to create active and control surfaces. The level of surface probe capture was recorded 4 min after the end of the injection as a frequency change.| Name | DNA sequence |
|---|---|
| ICP0 surface probe | 5′-Biotin-TCG CAT TTG CAC CTC GGC ACT CGG AGC G-3′ |
| ICP0 scrambled surface probe | 5′-Biotin-CCA TCG GCA TGT ACC GTA TCG GCG CGT C-3′ |
| ICP0 target sequence | 5′-CGC TCC GAG TGC CGA GGT GCA AAT-3′ |
| VP16 surface probe | 5′-Biotin-CTC GTT GGC GCG CTG AAG CAG GTT TTT G-3′ |
| VP16 scrambled surface probe | 5′-Biotin-ACC TGG GCA TGT ATG GTG TCG TCG CGT T-3′ |
| VP16 target sequence | 5′-AAA ACT TCC GTA CCC CTC AAA AAC CTG CTT CA-3′ |
| VP16 detection probe | 5′-GGG TAC GGA AGT TTT TCA CTC GAC-Biotin-3′ |
Hybridisation assay
Synthetic target ssDNA oligomers (ssDNA; TIB Molbiol, Berlin, Germany) were prepared in Tris buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, pH 7) (Table 1). Single-stranded target DNA at specified concentrations was injected over biotinylated probe-captured AKT♦iv Covalent chips for 3 min to allow hybridisation. After a dissociation period under Tris buffer flow at 25 µl min−1, surfaces were regenerated by two successive 30 s injections of 1 mM HCl. Frequency change due to hybridisation was recorded 180 s after the injection started.Signal enhancement assay
The running buffer used for the assay was Tris buffer comprising 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 0.05% Tween 20, pH 7. A sample of 10 mM biotin (biotin; Sigma-Aldrich, Poole, UK) in Tris buffer was injected for 1 min to block the remaining active sites of the NA layer. The VP16 target sequence was injected to the probe-immobilised sensor surface and allowed to hybridise for 3 min. Then VP16 detection probe at 0.5 or 2.5 µg ml−1 concentrations was injected to allow hybridisation with the VP16 target sequence for 3 min. Finally 5 µg ml−1 NA was injected to the resultant sensor surface to be captured by the VP16 detection probe for 3 min (Fig. 1). The frequency change due to oligomer hybridisation and NA capture were recorded 180 s after the injection started.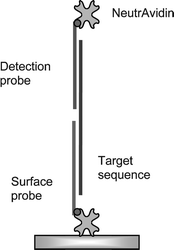 | ||
| Fig. 1 Schematic of the assay format for hybridisation signal enhancement by the capture of NA to the detection probe. | ||
Results and discussion
The RAP♦id 4 biosensor was used as the platform for the HSV type 1 nucleic acid tests. In order to select the most suitable sensor chip for nucleotide -based detection, three types of sensor chips – AKT♦iv Covalent, biotin, and CMD-T500 polymer chips – were compared. Reproducibility, signal-to-noise ratio, assay quality, percentage activity and limit of detection were investigated and compared for these three sensor chip types. After sensor chip selection, to reduce the limit of detection, a simple approach was applied by using NA as a secondary capture protein .HSV-ICP0 hybridisation assay
The ICP0 surface probe was captured onto the active flow cells of NA-immobilised AKT♦iv Covalent-, Biotin- and CMD-T500 polymer-sensor chips. The ICP0 scrambled surface probe was immobilised on separate flow cells as the control surfaces. Biotinylated 28-mer surface probe captured on NA-immobilised AKT♦iv Covalent chips produced an average frequency change of 116 ± 6 Hz; on the biotin chips the result was 27 ± 6 Hz; and on the CMD-T500 polymer chips 311 ± 51 Hz (n = 6 for all sensor chips).The specificity of the interaction was tested by employing a non-complementary probe on the sensor surface; the hybridisation signals resulting from these non-specific interactions were not more than 4 Hz (data not shown) for the AKT♦iv Covalent and biotin chips, and not more than 1 Hz (data not shown) for the CMD-T500 polymer chip. These results showed that the hybridisation on the sensor surfaces was specific and that the signal-to-noise ratio for CMD-T500 polymer chips was higher than the other sensor chips (Table 2). The non-specific response to the non-complementary sequence was subtracted from each hybridisation response by using Akubio Workbench Software.
| Sensor chip type | K D/µM | –dFmax/Hz | Signal/noisea | Surface activity (%)a | Z-Factora |
|---|---|---|---|---|---|
| a Results were calculated using ICP0 hybridisation data for 40 µg ml−1 target sequence (n = 6). | |||||
| Biotin | 0.021 ± 0.011 | 21 ± 6 | 6 | 73 | 0.65 |
| AKT♦iv Covalent | 0.018 ± 0.008 | 69 ± 14 | 20 | 61 | 0.89 |
| CMD-T500 polymer | 0.024 ± 0.004 | 118 ± 20 | 140 | 48 | 0.56 |
The calibration curve obtained with complementary oligonucleotides for the ICP0 surface probe in a concentration range between 0.3 and 40 µg ml−1 is shown in Fig. 2. The hybridisation responses were superimposed and aligned to the start of the sample injection and then fitted to a global 1 : 1 Langmuir binding model, giving a KD of ca. 0.021 µM for the CMD-T500 polymer, biotin and AKT♦iv Covalent chips (Table 2). Although the DNA capture and hybridisation response intensities varied among sensor chips, the KD values of oligonucleotide hybridisation were very similar to each other, indicating a good inter-assay consistency even with variant surface chemistry. The detection limit for target hybridisation was found to be ca. 9 × 10−8 M for all sensor chips.
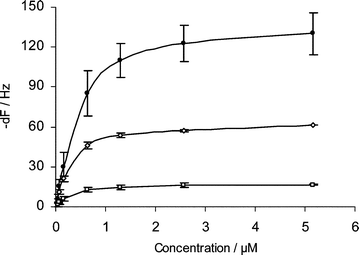 | ||
| Fig. 2 Frequency changes after 3 min of hybridisation of the ICP0 target sequence to the surface probe (n = 6). (●) CMD-T500 polymer chip, (◇) AKT♦iv Covalent chip, (□) biotin chip. | ||
Z-Factor analysis was employed to assess the quality of the assays using the different sensor chip types. The Z-factor18 provides an easy and useful measure for assay quality and has been a widely accepted standard. The Z-factor reflects both the assay signal dynamic range and the data variation associated with the signal measurements: a Z-factor between 0.5 and 1.0 indicates an excellent assay ; between 0 and 0.5 is marginal; and less than 0 means that the signals from the positive and negative controls overlap, indicating the invalidity of the assay results [eqn (1); average (µ) and standard deviation (σ) of both active and control DNA hybridisation results].
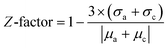 | (1) |
Z-Factor values were calculated for the hybridisation of 40 µg ml−1 ICP0 target sequence. The Z-factor values were above 0.5 for all sensor types indicating excellent assay performances; however, AKT♦iv Covalent chips with a Z-factor of 0.89 performed better than the other sensor chip types (Table 2).
The percent activities of the sensor chips were calculated from the ratio of the experimentally determined DNA hybridisation response with the theoretical maximum DNA hybridisation response. The activity for DNA hybridisation was higher for biotin chips (73%) than the other sensor chips, although the absolute magnitude of the hybridisation signal was lower (Table 2). This indicates potentially that whilst the biotin chip presents fewer binding sites to the solution phase for DNA capture, those sites present are better able to effect subsequent DNA hybridisation from solution.
Initial rate analysis is a useful tool to determine the concentration of unknown samples. The initial rates of hybridisation obtained from linear regression were concentration-dependent, and plots of initial rate against DNA concentration yielded a straight line that passed near the origin (Fig. 3). The standard deviations of the initial rate results for CMD-T500 polymer chips were much higher than the other sensor types; this would prevent accurate measurement of the concentration of unknown samples when using CMD-T500 polymer chips. Both AKT♦iv Covalent and biotin sensor chips showed very low standard deviations and a linear regression line with an R2 value of 0.99. The dynamic range of the initial rate correlation with DNA concentration was higher for AKT♦iv Covalent sensors than the biotin sensor chips.
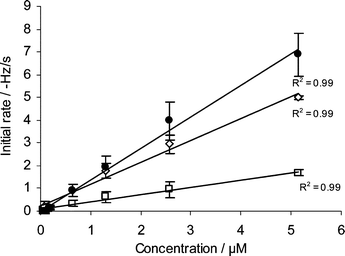 | ||
| Fig. 3 Initial rate results for ICP0 hybridisation (n = 6). (●) CMD-T500 polymer chip, (◇) AKT♦iv Covalent chip, (□) biotin chip. | ||
In summary, all three sensor types – AKT♦iv Covalent, CMD-T500 polymer and biotin chips – possessed a very similar limit of detection for ssDNA (9 × 10−8 M) and gave similar KD values for hybridisation (ca. 0.021 µM). The signal-to-noise ratio, Z-factor, percentage activity and initial rate values were calculated and compared for all sensor types. CMD-T500 polymer chips possessed a high signal-to-noise ratio; however, surface activity and Z-factor results were not as good as AKT♦iv Covalent sensor chips. In addition, the CMD-T500 assays possessed higher inter-assay variability. Although the activity of biotin chips was higher than the others, biotin chips had a very low signal-to-noise ratio and a smaller dynamic range with respect to the other sensor chip types. As such, both CMD-T500 and biotin chips were excluded from further optimisation studies, and AKT♦iv Covalent sensor chips were selected as the most suitable candidate for DNA hybridisation assays due to the higher assay quality dynamic range and reproducibility.
HSV-VP16 hybridisation and signal enhancement assay
50 µg ml−1 NA was immobilised to AKT♦iv Covalent sensors, then 10 µg ml−1 biotinylated probe was captured on all sensor surfaces. Biotinylated DNA capture on the NA layer resulted in a 243 ± 14 Hz response (n = 16, data not shown), followed by capping of the remaining NA active sites with 10 mM biotin.Two types of control assays were run to determine the non-specific binding of NA to the VP16 scrambled probe and to the VP16 surface probe. The VP16 scrambled sequences were captured onto the NA layer and this was followed by sequential injections of 1 µg ml−1 VP16 target sequence, 1 µg ml−1 VP16 detection probe and 5 µg ml−1 NA. The resulting non-specific binding of NA was 9 Hz (data not shown). As a second control, the VP16 surface probe was captured onto the sensor surface and then 5 µg ml−1 NA was injected. Non-specific binding of NA to the VP16 surface probe resulted in a 7 ± 1 Hz (n = 3) frequency change (Fig. 4).
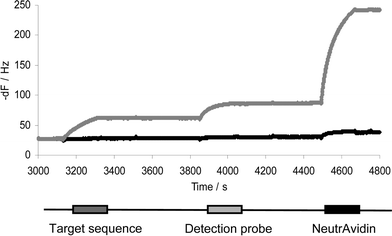 | ||
| Fig. 4 Sequential injections of 1 µg ml−1 target sequence, 1 µg ml−1 detection probe and 5 µg ml−1 NeutrAvidin to enhance the hybridisation signal. Grey: active flow cell; black: control flow cell. | ||
The hybridisation between the VP16 surface probe and the complementary oligonucleotide VP16 target sequence was tested in a concentration range varying from 5.2 × 10−11 to 1.3 × 10−7 M (Fig. 5). Whilst 5 and 10 ng ml−1 VP16 target sequence hybridisation to the VP16 surface probe did not result in any frequency response from VP16 detection probe hybridisation, subsequent NA capture on these surfaces resulted in 9 ± 3 and 17 ± 4 Hz responses respectively. The resultant theoretical detection limit of DNA hybridisation for this assay with NA signal amplification was 1 ng ml−1 (1 × 10−10 M). Notably, we have found in the course of this work that DNA hybridisation efficiency can be higher when hybridisation is performed at the annealing temperature in free solution rather than viain situ hybridisation to a probe on the biosensor surface. Hence, the first target sequence and detection probe were hybridised at 55 °C in a tube in free solution, then injected over the biosensor surface. Subsequent addition of NA resulted in a 5 ± 2 Hz frequency change for 5.2 × 10−11 M target concentration (Fig. 5). The same concentration of target sequence did not result in any response when hybridisation was performed on the sensor surface. In conclusion, the detection limit for VP16 target sequence hybridisation was 5.2 × 10−11 M when NA was used for signal enhancement and hybridisation performed at the annealing temperature.
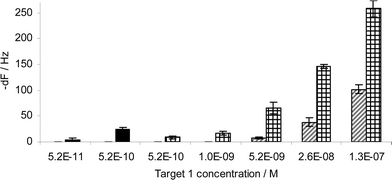 | ||
| Fig. 5 Diagonally striped columns: VP16 target sequence hybridisation onto the surface probe. Crossed columns: NeutrAvidin capture onto the VP16 detection probe. Black columns: NA capture after the injection of solution-hybridised VP16 target sequence and VP16 detection probe (n = 4 for all results). | ||
Conclusion
Nucleic acid -based testing has been increasingly used for the detection of viral pathogens. The implementation of biosensors to nucleic acid -based recognition can provide real-time, label-free and quick testing of samples. Current optical label-free biosensors are large and their detection limits require significant improvement, especially in complex media typical of clinical matrices. Piezoelectric biosensors are able to operate in un-diluted, un-processed complex media10 and, being entirely electronic, are suitable for miniaturisation. A preliminary study was conducted for the detection of viral sequences using a piezoelectric biosensor , the RAP♦id instrument. The hybridisation performance of three different sensor chips including AKT♦iv Covalent chips, biotin chips and CMD-T500 polymer chips were compared and AKT♦iv Covalent chips were found to be the most suitable candidates for further studies – due to their hybridisation capacity, high reproducibility and higher dynamic range. After a hybridisation time of only three minutes and NA capture as a signal enhancer, it was possible to detect HSV viral nucleic acids at 5.2 × 10−11 M concentration. This level of sensitivity necessitates amplification of DNA by means of PCR or PCR -like amplification methods; however, by reducing the detection limit of the DNA to the region of 10−18 M or lower, it may be possible to avoid PCR amplification. Further studies will continue to improve the limit of detection for the recognition of HSV type 1 and other viral nucleic acids .Acknowledgements
M. A. C. and Y. U. would like to express their gratitude to the National Institute of Allergy and Infectious Diseases for partial financial support, NIH Grant Number AI-061243-02.References
- M. J. Espy, J. R. Uhl, P. S. Mtchell, J. N. Thorvilson, K. A. Svien, A. D. Wold and T. F. Smith, J. Clin. Microbiol., 2000, 38, 795 CAS.
- N. C. Fawcett, R. D. Craven, P. Zhang and J. A. Evans, Anal. Chem., 1998, 70, 2876 CrossRef CAS.
- S. Tombelli, M. Minunni, A. Santucci, M. M. Spiriti and M. Mascini, Talanta, 2006, 68, 806 CrossRef CAS.
- S. Efstathiou and C. M. Preston, Virus Res., 2005, 111, 108 CrossRef CAS.
- R. Hagglund and B. Roizman, J. Virol., 2004, 78, 2169 CrossRef CAS.
- T. J. Wu, G. Monokian, D. F. Mark and C. R. Wobbe, Mol. Cell. Biol., 1994, 14, 3484 CAS.
- M. H. Hancock, J. A. Corcoran and J. R. Smiley, Virology, 2006, 352, 237 CrossRef CAS.
- M. E. Napier, C. R. Loomis, M. F. Sistare, J. Kim, A. E. Eckhardt and H. H. Thorp, Bioconjugate Chem., 1997, 8, 906 CrossRef CAS.
- P. Kara, B. Meric, A. Zeytinoglu and M. Ozsoz, Anal. Chim. Acta, 2004, 518, 69 CrossRef CAS.
- K. A. Marx, Biomacromolecules, 2003, 4, 1099 CrossRef CAS.
- S. Bruckenstein and M. Shay, Abstr. Pap. Am. Chem. Soc., 1988, 196, 132.
- T. Jiang, M. Minunni, P. Wilson, J. Zhang, A. P. F. Turner and M. Mascini, Biosens. Bioelectron., 2005, 20, 1939 CrossRef CAS.
- F. Mannelli, A. Minunni, S. Tombelli, R. H. Wang, M. M. Spiriti and M. Mascini, Bioelectrochemistry, 2005, 66, 129 CrossRef.
- T. Liu, J. Tang and L. Jiang, Biochem. Biophys. Res. Commun., 2004, 313, 3 CrossRef CAS.
- I. Willner, F. Patolsky, Y. Weimann and B. Willner, Talanta, 2002, 56, 847 CrossRef CAS.
- Y. Weizmann, F. Patolsky and I. Willner, Analyst, 2001, 126, 1502 RSC.
- X. Li, K. S. J. Thompson, B. Godber and M. A. Cooper, Assay Drug Dev. Technol., 2006, 4, 565 CrossRef CAS.
- J. H. Zhang, T. D. Y. Chung and K. R. Oldenburg, J. Biomol. Screen., 1999, 4, 67 CrossRef.
Footnote |
| † © Akubio Limited (2007). Akubio, the Akubio acoustic biosensors logo, and the diamond motif are registered trademarks of Akubio Ltd; RAP, RAP♦id, AKT♦iv, LINK♦it are trademarks of Akubio Ltd. |
| This journal is © The Royal Society of Chemistry 2008 |