Is there a future for sequential chemical extraction?†
Jeffrey R.
Bacon
a and
Christine M.
Davidson
*b
aThe Macaulay Institute, Craigiebuckler, Aberdeen, UK AB15 8QH
bWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, UK G1 1XL. E-mail: c.m.davidson@strath.ac.uk
First published on 26th September 2007
Abstract
Since their introduction in the late 1970s, sequential extraction procedures have experienced a rapid increase in use. They are now applied for a large number of potentially toxic elements in a wide range of sample types. This review uses evidence from the literature to consider the usefulness and limitations of sequential extraction and thereby to assess its future role in environmental chemical analysis. It is not the intention to provide a comprehensive survey of all applications of sequential extractions or to consider the merits and disadvantages of individual schemes. These aspects have been covered adequately in other, recent reviews. This review focuses in particular on various key issues surrounding sequential extractions such as nomenclature, methodologies, presentation of data and interpretation of data, and discusses typical applications from the recent literature for which sequential extraction can provide useful and meaningful information. Also covered are emerging developments such as accelerated procedures using ultrasound- or microwave energy-assisted extractions, dynamic extractions, the use of chemometrics, the combination of sequential extraction with isotope analysis, and the extension of the approach to non-traditional analytes such as arsenic, mercury, selenium and radionuclides.
 Jeffrey R. Bacon | Jeffrey Bacon is a senior researcher at the Macaulay Institute in Aberdeen with a particular interest in the use of mass spectrometry to study radiogenic isotopes in the environment. He was involved in the development of the BCR sequential extraction procedure and participated in the BCR reference material programme. |
 Christine M. Davidson | Christine Davidson is a senior lecturer in analytical chemistry at the University of Strathclyde, specialising in development of methods for measurement and fractionation of potentially toxic elements. Her BSc and PhD degrees are from the University of Glasgow, with post-doctoral experience in Utrecht, Netherlands, and at SURRC, UK. |
1. Introduction
The environmental behaviour of potentially toxic elements (PTEs) depends critically on the form in which they occur.2 The manner in which an element is bound to the solid components of environmental solids, such as soils or sediments, influences the mobility and, ultimately, the bioavailability and toxicity of the element to organisms. As a result there is considerable interest in improving the understanding of element–solid phase associations in natural and polluted systems.Direct determination of the chemical forms of trace elements in environmental samples such as soils can be achieved by means of various instrumental techniques,3 notably synchrotron-based X-ray radiation fluorescence (SXRF),4,5 particle-induced X-ray emission (PIXE),5 X-ray absorption near edge structure (XANES),6 and extended X-ray absorption fine structure (EXAFS)4,5spectroscopy. Although powerful, these techniques are not widely available and may offer poor detection limits, meaning that they can be applied only to heavily contaminated samples.7–10
Hence, over the past three decades, interest has increased markedly in the use of indirect approaches such as sequential chemical extraction (Fig. 1). In sequential extraction, a series of reagents is applied to the same sample to sub-divide the total metal content. The ‘vigour’ of the treatment generally increases through the steps of the procedure, from initial mild conditions (e.g. shaking with water, a salt solution or dilute acetic acid) to the use of much harsher reagents (e.g. hot mineral acid). The PTEs extracted early in the process are thus generally those most weakly bound to the solid phase. Hence, they have greater potential mobility, and environmental impact, than those released later.
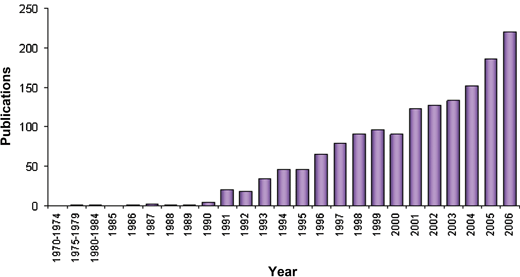 | ||
| Fig. 1 Growth in publications featuring the use of sequential extraction for fractionation of trace metals (Source: ISI Web of Science; search parameters: ‘sequential extraction’ AND ‘meta*’). | ||
In 1991, Ure11 defined chemical speciation as either ‘the active process of identification and quantification of the different defined species forms or phases in which an element occurs in a material’ or ‘the description of the amounts and kinds of species, forms or phases present in the material’. He proposed that speciation be divided into three classes:
(a) Classical speciation refers to specific chemical compounds or oxidation states of elements, e.g. cerussite (PbCO3) vs.pyromorphite [Pb5(PO4)3Cl]; CrIII vs. CrVI.
(b) Functional speciation refers to the observed role or behaviour of the element, and is characterized by terms such as ‘plant available’ or ‘mobile’ species.
(c) Operational speciation refers to the situation where the reagent used to extract the sample defines the species, e.g. ‘acetic acid soluble’ or ‘moderately reducible’ species.
Sequential chemical extraction is an example of operational speciation.
Later, IUPAC12 published a definition of speciation which distinguished the process of identifying species (‘speciation analysis’) from the description of the species themselves. The IUPAC definition of speciation corresponds roughly to the ‘classical’ definition of Ure. Hence, although the term ‘operational speciation’ is still widely used, it is more correct to refer to sequential extraction as a method for ‘fractionation’ of PTEs.
To quote an IUPAC report13 on the determination of trace elements bound to soils and sediment fractions, “despite some drawbacks, the sequential extraction method can provide a valuable tool to distinguish among trace element fractions of different solubility related to mineralogical phases. The understanding of the speciation of trace elements in solid samples is still rather unsatisfactory because the appropriate techniques are only operationally defined”. This is the nub of the problem in using sequential extractions. The ever increasing use of sequential extractions indicates that this approach is thought to provide meaningful and useable information and yet in many papers no real attempt is made to interpret the data other than to say that so much metal is associated with a certain phase. There appear to be many misconceptions in existence, even to the meaning of ‘operationally defined’, which can lead to confusion. The aim of this review is to outline the development and current status of chemical sequential extraction and to highlight some of the issues that still exist in the application of the procedure and interpretation of results.
2. History
The growth in interest in sequential extraction may be traced back to the classic work of Tessier, Bison and Campbell in 1979.14 They used a five-stage extraction (Table 1) to fractionate cadmium, cobalt, copper, iron, lead, manganese, nickel and zinc in river sediments containing low levels of PTEs. The reagents used were selected on the basis of their ability to remove analytes from specific, major, sediment phases – either by exchange processes or by dissolution of the target phase. Extraction steps also corresponded with, or at least represented extremes of, important changes in environmental conditions that could affect metal binding in sediments: acidification (e.g. in response to an input of acidified rainwater or industrial discharge); reduction (e.g. as may occur following post-depositional burial in a sediment column) and oxidation (e.g. as might occur following dredging and land-deposition of anoxic sediments).| Tessier (ref. 14) | Revised BCR (ref. 21) | |||||
|---|---|---|---|---|---|---|
| Reagent | Fraction label and nominal target phase(s) | Reagent | Fraction label | Nominal target phase(s) | ||
| a Although not officially a step in the sequential extraction, it is recommended that the residue at the end of Step 3 be digested with aqua regia and the sum of the four fractions be compared with the results of a separate aqua regiadigestion of the sample. | ||||||
| Step 1 | 1.0 mol l–1MgCl2 at pH 7.0 | Exchangeable | Step 1 | 0.11 mol l–1 CH3COOH | Exchangeable, water- and acid-soluble | Soluble and exchangeable cations, and carbonates |
| Step 2 | 1.0 mol l–1 CH3COONa adjusted to pH 5 with CH3COOH | Bound to carbonates | ||||
| Step 3 | 0.04 mol l–1 NH2OH·HCl in 25% CH3COOH (96 °C) | Bound to Fe–Mn oxides | Step 2 | 0.5 mol l–1 NH2OH·HCl at pH 1.5 | Reducible | Fe–Mn oxyhydroxides |
| Step 4 | HNO3/H2O2 (85 °C) then 3.2 mol l–1 CH3COONH4 in 20% HNO3 | Bound to organic matter and sulfides | Step 3 | H2O2 (85 °C) then 1.0 mol l–1 CH3COONH4 | Oxidisable | Organic matter and sulfides |
| Step 5 | HClO4/HF | Residual | (Step 4)a | Aqua regia | Residual | |
Sequential extraction was thus originally developed to provide information on potential impacts of sediment-bound PTEs on water quality. However, application to soil soon followed15,16 and, by the early 1990s a number of researchers were using the approach to fractionate PTEs (and, in some cases, radionuclides) in a variety of substrates. The use of different procedures, with different numbers of steps, reagents and extraction conditions, meant that it quickly became difficult to draw meaningful comparisons between results obtained in different laboratories. The need for standardization became clear.
The Community Bureau of Reference of the Commission of the European Communities (BCR) commissioned research which led to the development of a harmonized, three-stage, sediment sequential extraction protocol.17,18 The principal difference in this new scheme, with respect to that of Tessier, was that the first two steps of the Tessier scheme were replaced by a single step. In addition, larger sample amounts and extractant volumes were used to allow more representative sampling and to avoid some of the analytical difficulties associated with the use of small extractant volumes. In light of increasing concerns over the specificity of reagents used in sequential extraction procedures (see Section 3), the fractions were labeled according to chemical processes rather than target mineral phases.
Problems were reported with irreproducibility of, in particular, Step 2 of the original BCR procedure (see, for example, ref. 19) and, after a thorough re-evaluation in the late 1990s,20 a revised protocol was recommended21 (shown in Table 1). Recognizing the need for improved quality control in sequential extraction, the BCR also led developments such as the production of certified reference materials (CRMs).22 Sediments certified for amounts of analytes extractable by both original23,24 and revised25,26BCR sequential extraction protocols were produced (see Table 2). It was also recommended that, when using the revised BCR protocol, an additional step (aqua regiadigestion of the residue from Step 3) be performed and the sum of the four steps of the sequential extraction be compared with the result of a separate aqua regiadigestion of the whole soil (pseudototal content). In this way the overall effectiveness of the sequential extraction process and element recoveries can be assessed.
| BCR CRM 601 (metals extractable by the revised BCR procedure)d | |||||
|---|---|---|---|---|---|
| Cd | 4.45 ± 0.67c | 3.95 ± 0.53c | 1.91 ± 1.43c | 1.3 ± 2.2c | 11.5 ± 1.9c |
| Cr | 0.35 ± 0.08c | 10.6 ± 0.9c | 14.4 ± 2.6c | 78.2 ± 6.5c | 112 ± 9.5c |
| Cu | 10.5 ± 0.8c | 72.8 ± 4.9c | 78.6 ± 8.9c | 60.4 ± 4.9c | 230 ± 15c |
| Ni | 7.82 ± 0.84c | 10.6 ± 1.2c | 6.04 ± 1.27c | 50.5 ± 4.3c | 78.8 ± 6.7c |
| Pb | 2.28 ± 0.44c | 205 ± 11c | 19.7 ± 5.8c | 38.0 ± 8.7c | 288 ± 52c |
| Zn | 261 ± 13c | 266 ± 17c | 106 ± 11c | 161 ± 14c | 833 ± 17c |
| BCR CRM 701 (metals extractable by the revised BCR procedure)e | |||||
|---|---|---|---|---|---|
| a Uncertainties quoted are half widths of the 95% confidence intervals of the mean values. b Aqua regia-soluble PTE content. c Indicative value. d Uncertainties quoted are standard deviations (n = 7 for Steps 1–3, n = 6 for Step 4 and pseudototal). e Uncertainties quoted are half widths of the 95% confidence intervals of the mean values for Steps 1–3, but standard deviations for Step 4 and pseudototal values. | |||||
| Cd | 7.34 ± 0.35 | 3.77 ± 0.28 | 0.27 ± 0.06 | 0.13 ± 0.08c | 11.7 ± 1.0c |
| Cr | 2.26 ± 0.16 | 45.7 ± 2.0 | 143 ± 7 | 62.5 ± 7.4c | 272 ± 20c |
| Cu | 49.3 ± 1.7 | 124 ± 3 | 55.2 ± 4.0 | 38.5 ± 11.2c | 275 ± 13c |
| Ni | 15.4 ± 0.9 | 26.6 ± 1.3 | 15.3 ± 0.9 | 41.4 ± 4.0c | 103 ± 4c |
| Pb | 3.18 ± 0.21 | 126 ± 3 | 9.3 ± 2.0 | 11.0 ± 5.2c | 143 ± 6c |
| Zn | 205 ± 6 | 114 ± 5 | 45.7 ± 4.0 | 95 ± 13c | 454 ± 19c |
Since the early 1990s, sequential extraction has continued to increase in popularity. In addition to the sediments originally envisaged, the approach has been applied to a wide variety of substrates including agricultural soils,27,28 soils amended with organic wastes,29–33 rhizosphere soils,34–39 urban soils,40,41 forest soils,42,43 industrial (contaminated) soils,44,45 urban sediments (road particulates),46 mine spoil,47–49 sewage sludge,50–52 composts,53–56 incinerator ashes,57 medical waste fly ash,58 airborne particulate matter,59,60 electric arc furnace dust61 and gas pipeline corrosion products.62 Although a large number of different protocols have been reported, the Tessier and BCR schemes remain amongst the most widely used. A comprehensive review of sequential extraction schemes was provided in 2002 by Filgueiras, Lavilla and Bendicho.63 The review of Young et al.64 is recommended for providing a clear introduction to the development of sequential extractions, some of the limitations such as lack of specificity and some of the recent innovations for improving the procedure. Other reviews have included an overview of the use of leaching/extraction tests for risk assessment of trace metals in contaminated soils and sediments,65 the use of sequential extraction procedures for the characterization and management of contaminated soils,66 the fractionation of metals in atmospheric aerosols67 and recent methodological advances, in particular for on-line dynamic fractionation.68
An ongoing limitation to the use of sequential extraction has been the availability of only a few reference materials for checking the performance of methods and laboratories. Various authors have therefore attempted to increase the range available by applying standard69–80 or other81,82 procedures to generate indicative extractable metal concentrations in additional reference materials, typically soils or sediments already certified for their total PTE contents. This has provided useful information (see Table 3). However, such data should be used with care since they are generally generated in a single laboratory and, therefore, not subjected to the same degree of inter-laboratory assessment as occurs during certification of a new reference material. Further work in this area will, however, refine the reliability of published results78 and is therefore to be encouraged.
| CRM | Type of material | Procedure | PTEs measured | Reference |
|---|---|---|---|---|
| NIST SRM 2709 | Relatively uncontaminated agricultural soil | Tessier | Al, Ca, Cd, Co, Cu, Fe, K, Mn, Ni, (P), Pb, Sr, Ti, V, Zn | 69 |
| NIST SRM 2710 | Highly contaminated pasture soil | Tessier | Al, Ca, Cd, Co, Cu, Fe, K, Mn, Ni, (P), Pb, Sr, Ti, V, Zn | 69 |
| Original BCR | Cd, Cu, Pb, Zn | 70 | ||
| Revised BCR | Al, Cu, Fe, Mn, Pb, Zn | 72 | ||
| Revised BCR | Al, Cu, Fe, Mn, Pb, Zn | 75 | ||
| Revised BCR | Al | 77 | ||
| NIST SRM 2711 | Moderately contaminated agricultural soil | Tessier | Al, Ca, Cd, Co, Cu, Fe, K, Mn, Ni, (P), Pb, Sr, Ti, V, Zn | 69 |
| Original BCR | Cd, Cu, Pb, Zn | 70 | ||
| Revised BCR | Al, Cu, Fe, Mn, Pb, Zn | 72 | ||
| Revised BCR | Al, Cu, Fe, Mn, Pb, Zn | 75 | ||
| Revised BCR | Al, As, Cd, Co, Cr, Cu, Fe, Mn, Ni, Pb, Sb, Zn | 78 | ||
| Revised BCR | Cd, Cu, Fe, Mn, Pb, Zn | 79 | ||
| NIST SRM 1648 | Urban air particulate matter | Original BCR | Cd, Cu, Fe, Mn, Zn | 73 |
| BCR CRM 483 | Sewage sludge-amended soil | Revised BCR | Cd, Cr, Cu, Ni, Pb, Zn | 71 |
| Revised BCR | Al, Cu, Fe, Mn, Pb, Zn | 72 | ||
| Revised BCR | Cu, Fe, Mn, Pb, Zn | 74 | ||
| Revised BCR | Al, Cu, Fe, Mn, Pb, Zn | 75 | ||
| Revised BCR | Cd, Cr, Cu, Ni, Pb, Zn | 76 | ||
| CW 7 | Dust from ventilation shaft of a road tunnel | Revised BCR | Al, Cu, Fe, Mn, Pb, Zn | 72 |
| Revised BCR | Al | 77 | ||
| CANMET RSS SO-2 | Ferro-humic podzol soil | Revised BCR | Al | 77 |
| CANMET RSS SO-4 | Black soil | Revised BCR | Al | 77 |
| HPS CRM SA-B | Sandy soil | Revised BCR | Al | 77 |
| WEPAL SRM RTH 912 | Loess soil | Revised BCR | Al | 77 |
| RTC NMCRM 025–050 | Moderately contaminated soil | Revised BCR | Al | 77 |
| GBW 07401 | Chinese soil | Original BCR | Tl | 80 |
3. Scope and limitations of sequential extraction
Sequential extraction is thus now widely accepted and adopted. The approach has led to improved understanding of the behaviour of PTEs in environmental samples, and generated large amounts of data useful in assessing potential risks from environmental contaminants. However, along with the proliferation in applications have grown misconceptions about the significance of the results obtained.83 Procedures are often applied uncritically84 and, in particular, the operational nature of the extraction procedure is frequently ignored (or – worse – noted but then disregarded) by authors. This issue is discussed in more detail in Section 4.It is extremely important to appreciate that sequential extraction only divides the PTE content of a test sample into portions soluble in particular reagents under particular conditions. Whilst these reagents are often selected with the intention that they should target well-defined mineral phases – and may indeed do so in many cases – such specificity cannot be guaranteed. Hence, interpretation of the results of sequential extraction in terms of binding of trace metals to specific minerals is unjustifiable, unless additional, X-ray-based, analytical techniques are applied to the residues at each stage in the extraction to identify precisely the solid components remaining.84–93
Schemes continue to be described which are claimed to target specific, well-defined, phases successfully. Poulton and Canfield94 developed a scheme to target seven ‘operationally derived’ iron pools in sediment samples and validated the specificity on grain-size-separated sediments. An improved procedure using hydroxylamine hydrochloride and acidified hydrogen peroxide, also used in both the BCR and Tessier schemes, was developed in order to dissolve specifically manganese oxide phases without any significant dissolution of iron oxide phases.95
Such papers are, however, heavily outnumbered by papers which report clear evidence for the non-specificity of sequential extraction procedures. The comparison by Parat et al.96 of three different procedures provided evidence for lack of specificity and the operational nature of the extractions. Sodium acetate, generally defined as extracting exchangeable metals or carbonate-bound metals, could remove considerable amounts of metals in forms other than exchangeable ones. Copper distribution was affected by the position of the oxidation step in the sequence of extraction steps. In a similar comparison, Tokalioglu et al.97 also concluded that the amount of metal released at each step of the leaching procedure depended both on the type of reagents used and the sequence in which they were applied. As the measured amount of metal associated with a particular phase is strongly dependent on the extractant and procedure used, it has been suggested that at least two independent procedures should be used.98 In most cases this would be considered impractical. Hanahan99 found that sodium acetate could also release metals associated with hydroxide minerals. Independent studies of mining wastes100 and anoxic sediments101,102 demonstrated that hydroxylamine hydrochloride can dissolve sulfide minerals so sulfide-bound metals could mistakenly be interpreted as being bound to iron oxide species. Dermatas et al.103 concluded that lead in the soils of firing ranges could be extracted in any step of the procedure depending on the soil buffering capacity even though scanning electron microscopy revealed 95% of the lead to be in carbonate or oxide forms. Perhaps the most extreme demonstration of the dependency of interpretation on the procedure used was the finding by Doelsch et al.104 that the amendment of a tropical soil with sewage sludge led to an increase of metals associated with the reducible fraction according to one sequential extraction scheme but to a decrease according to another scheme.
A number of other problems have been identified that cannot be explained by specific causes. Mostly unreliable results were obtained for chromium using 11 different schemes,105 serious discrepancies were found for manganese using an ‘optimized’ BCR procedure,75 no reliable data for nickel in unpolluted soils could be obtained using the BCR procedure106 and systematic under-recovery was observed for a seven-step procedure.107 In comparison to single extractant procedures, sequential extraction was considered more aggressive and gave higher extractable concentrations.108,109
Although not specifically a problem of sequential extraction but more of speciation studies in general, thought should be given to the effects of sample preparation on metal distribution, in particular in the study of sediments. Sample drying and grinding of sediments has consistently been shown to lead to re-distribution of the metals to more extractable forms.110–112 Unfortunately, the reproducibility of analysis on wet samples was considerably poorer than that for dried samples.112Freeze-drying did not preserve metal partitioning in anoxic sediment cores.113 It has been suggested that sediment and soil samples should be sampled and analysed under an inert atmosphere in order to reduce the risk of oxidation.114
There are several reasons why sequential extraction does not determine quantitatively the trace metals associated with specific mineral phases in environmental solids. These include:
(a) re-distribution of analytes among phases during extraction;
(b) non-selectivity of reagents for target phases;
(c) incomplete extraction;
(d) precipitation of ‘new’ mineral phases during extraction.
A considerable body of evidence has accumulated confirming the importance of these effects. This evidence is based on a large number of studies which fall into two groups – those that have utilized pure and synthetic substrates and those that utilized ‘real’ samples.
3.1 Pure and synthetic substrates
Even before publication of the Tessier sequential extraction, Guy et al.115 had demonstrated that low analyte recoveries were obtained when attempting to sequentially extract simple binary mixtures of bentonite, MnO2 and humic acid that had been spiked with Cu and Pb. This poor performance was attributed to either post-extraction re-adsorption of analytes on residual solids or incomplete dissolution of the target phase. Many studies conducted on model sediments116,117 and soils118–120 have since confirmed that significant re-distribution of analytes occurs during both Tessier116,119,120 and BCR117,118 protocols.Work on sequential extraction of seawater-spiked mineral phases121 illustrated the non-specificity of the BCR procedure with, for example, the majority of iron oxyhydroxide-bound metals being released in Step 1, rather than in Step 2 (the reduction step) as expected. Non-selectivity was also observed during application of the Tessier extraction to spiked soil components.120,122 Whilst good recoveries of Cu, Pb and Zn (generally >90%) could be obtained when individual components were subjected to a single extraction with their corresponding reagent (indicated in Table 1), significant amounts were also released when reagents were applied to non-target phases. For example, although intended to release ‘metals bound to carbonates’, it was found that acidified sodium acetate could also remove around 50% of analytes associated with Fe/Mn oxides.120
Premature extraction of organically-bound metals has been noted in both the Tessier122 and the BCR procedures,117,121 and presumably occurs because analytes can be liberated by exchange processes as well as following destruction of the organic matter. It is thus clear that neither Step 4 of the Tessier procedure, nor Step 3 of BCR, can be considered accurately to represent the entire pool of ‘metals bound to organic matter’.
3.2 Environmental substrates
The validity of extrapolating results obtained for synthetic substrates – or based on the use of high spike concentrations – has been questioned.123 However, many authors have now reported similar findings for ‘real’ samples. Re-distribution of lead from manganese oxide to iron oxide phases was shown to occur when samples of a naturally precipitated mixed oxide from a former lead mine were treated with hydroxylamine hydrochloride.85 Sequential extraction combined with EXAFS analysis of mine tailings demonstrated significant re-adsorption of lead liberated by Tessier Step 1.10 This inability of 1.0 mol l–1MgCl2 to retain added analytes in soluble forms was also noted by Gomez-Ariza et al.124 when using a ‘standard additions’ approach. They also showed that the degree of (re-)adsorption depended critically on the geochemical characteristics of the sediment studied.Radiotracers have provided further insight into the behaviour of PTEs undergoing sequential extraction. Radionuclides have the advantage that they can be added in very small quantities that do not alter significantly the total analyte concentrations in the sample. When aliquots of 109Cd, 65Zn and 212Pb were added at the start of Step 1 of the BCR extraction of a CRM, it was found that only 20–30% of copper and zinc,125 but 60–90% of lead,126 were scavenged from the acetic acid solution by the (solid) reducible fraction. When 212Pb was instead added to the reagent at the start of BCR Step 2, 35–85% of the activity partitioned into the solid phase but, when added at the start of BCR Step 3, the tracer remained in solution.126
Incomplete dissolution of carbonates during Step 1 of the BCR protocol has been demonstrated by X-ray analysis of the residue at the end of the extraction step, for both urban canal sediment86 and soil.84 The latter study focused on a calcareous soil, where the amount of acetic acid added was insufficient to dissolve the ca. 27% dolomite present. Carbonate-bound metals were not recovered until Step 2 or even Step 3 of the procedure, and the increased pH of the extract – resulting from neutralization of the acid – interfered with the partitioning of elements in subsequent steps (an effect also observed in carbonate-rich urban soils).41 The ability of a sample to alter (raise) markedly the initial pH of an added extractant, with subsequent decrease in solubility of extracted analytes, was also noted by Bermond in a detailed investigation of the role of H+ in sequential extraction,127 and was one of the factors that led to revision of the original BCR extraction.20
Enhanced dissolution efficiency can sometimes be achieved by repeating an extraction step before progressing to the next,84,128,129 and it has been suggested that use of such replicate extractions, together with careful monitoring of the pH,84,129 major element content,129 or redox potential129 of the extracts, can be a useful strategy.
Extraction yields are also low if the ratio of volume of extractant to mass of sample (v : m) is too low.130 The recommendation that large extractant volumes should be used has implications, however, for analytical detection of the extracted metals. It is important to define clearly a constant v : m ratio as extractability of metals varies with the ratio, highlighting the operational nature of the procedure.131
The re-distribution of PTEs can also result from the formation, during the extraction process, of new mineral phases not originally present in the sample. This can make results extremely difficult to interpret. For example, Zhu et al.132 interpreted fractionation data, obtained following the addition of a variety of phosphate amendments to soil, as indicating that such treatments could transform lead from non-residual to residual forms, thus successfully reducing the potential for plant uptake of this PTE. However, Scheckel et al.90,133 demonstrated that, in heavily phosphate-amended soil, insoluble lead phosphate (pyromorphite) was formed only during the sequential extraction process itself.
Artifacts can also originate from reagents applied. Significant amounts of aluminium, iron and lead were extractable from forest soils under reducing conditions but this fraction would be underestimated if sodium pyrophosphate were used in a previous step.134 Lead mobility in forest soils could therefore also be underestimated. Another example is the precipitation of insoluble oxalates following use of acidified ammonium oxalate (Tamm's reagent).86,135
It is well known that soils and sediments do not contain discrete particles of different minerals, but consist of complex mixtures of components.115,116 This means that, even where a reagent is completely specific, the intended target mineral phase could be inaccessible due, for example, to occlusion within a dissimilar mineral coating.136 Whilst this problem may be overcome by grinding the sample finely, grinding will expose new faces of solid components and increase the surface area for leaching, meaning that the data subsequently generated by sequential extraction may no longer reflect accurately the likelihood of PTE mobilization under the original environmental conditions.
4. Issues
4.1 Nomenclature
It is perhaps surprising that there is no commonly accepted abbreviation for sequential extractions. This, together with the fact that SE should not be used as it is already used for supercritical extraction, has resulted in a number of different abbreviations being used for essentially the same thing. These abbreviations include SCE (sequential chemical extraction), SEP (sequential extraction procedure), SES (sequential extraction scheme), SET (sequential extraction test), SSD (selective sequential dissolution), SSE (both selective sequential extraction and sequential selective extraction) and SSEP (short sequential extraction procedure).No official distinction is made between the two BCR procedures which exist. Papers frequently refer to ‘the BCR’ method or protocol but rarely to the specific procedure being employed. Only by looking at the detail in the method description can the reader determine which method was used. Confusion is greatest when authors refer to one of the two methods but then present details for the other method. It is not unknown for authors to state that they have used ‘the BCR method’ and to give references to both methods. It remains a mystery as to which one they actually used. Some authors do distinguish between the two methods by referring to them as the ‘original’ and ‘revised’ or ‘modified’ methods. Even this can lead to confusion. Does a reference to a revised BCR procedure mean that the authors have used the officially revised scheme or that they have themselves altered one of the two BCR procedures to meet their own requirements? One can in fact have a modified ‘revised BCR procedure’. Confusion could be reduced greatly if there were some generally accepted way of distinguishing between the two procedures. Simple ways would be to redefine the BCR procedures as the ‘BCR (1993)’ and ‘BCR (1999)’ schemes or as the ‘BCR1’ and ‘BCR2’ schemes.
Why is this important? There is increasing evidence that the two BCR procedures can produce very different results, in particular for lead. Mossop and Davidson,74 Sutherland and Tack137 and Bacon et al.138 have all compared the two BCR procedures and found significant differences between the two methods. It could be concluded that the modifications incorporated into the second procedure had been successful in attacking more completely the oxide phases but for lead the order of magnitude shift from the ‘oxidisable’ fraction to the ‘reducible’ fraction leads to severe problems of interpretation. This is best illustrated by the study of Jensen et al.139 in which the ‘original’ BCR procedure was used to study the speciation of lead in industrially polluted soils. Most lead was extracted in the ‘oxidisable’ fraction and it was concluded that lead adsorbed preferentially to organic matter. The conclusions would probably have been quite different had the ‘revised’ BCR procedure been used.
These findings emphasize the operational nature of the procedure. There appears to be growing awareness that sequential extraction procedures are operationally defined and that the results cannot be interpreted as metals being bound to specific phases. Reference to specific phases continues to happen, however, and, more confusingly, mixed terminology is frequently used. It is not unusual to read sentences like “The association of heavy metals with operationally defined solid phase fractions (carbonates, iron and manganese oxides, sulfides/organics and residual) was assessed”. What is the message that this gives? That the procedure, as defined by the operations carried out, targets the specific phases listed successfully? Or that the procedure is defined by the operations carried out and that the names given to each fraction are the nominally targeted phases and used for convenience rather than accuracy?
Most authors probably use the names of specific phases as a matter of convenience. The discussion of data is, however, frequently based on association of metals with specific phases. It is difficult to find a terminology which is accurate and yet simple to use. Relatively few papers have followed the examples set by Kersten et al.140 who referred to ‘exchangeable’, ‘reducible’, ‘oxidisable’ and ‘residual’ fractions, or Gobeil et al.141 who referred to ‘acetate buffer extractable Pb’ and ‘hydroxylamine/acetic acid extractable Pb’. The BCR method has never been presented as targeting specific phases and terms such as ‘reducible’ and ‘oxidisable’ fractions are preferred. This, however, is still rather vague even though probably the best manageable option. The differences between the two BCR procedures highlight the fact that the ‘reducible’ fraction, as defined by the original BCR procedure, is different from the ‘reducible’ fraction as defined by the revised BCR procedure. To be accurate one would have to refer to the ‘fraction reducible in 0.1 M hydroxylamine hydrochloride at pH 2’ or the ‘fraction reducible in 0.5 M hydroxylamine hydrochloride at pH 1.5’. This is clearly very clumsy and impractical. Perhaps the only solution is to define the fractions in the Methods section by the operations used and to refer to them simply as fraction 1, 2, 3, etc. Phrases such as ‘operationally defined Fe and Mn oxides (reducible fraction)’ are best avoided.
4.2 Methodology
Although authors frequently cite, for example, the BCR method as the procedure they used, close inspection can reveal that there are differences between the conditions as specified by the BCR procedure and those described by the authors. This applies in particular to the shaking conditions. Researchers can only use equipment they have available so, combined with a common lack of understanding of the term ‘end-over-end shaker’, extractions can be performed under a range of conditions. Jensen et al.,139 for example, followed the BCR procedure yet used a shaking speed of 100 rpm (type of shaker unspecified). Not all laboratories have air conditioning so extractions carried out in southern Europe will probably be outside the temperature range specified by the BCR procedure. These divergencies from the specified procedures are rarely recorded.Do these divergences matter? Probably not, but the true answer is that we don't know because these aspects have not been thoroughly investigated. The energy being placed into the shaking process could well influence the extraction efficiency but most shaking is carried out overnight so the extraction should be complete in that timescale. If too much energy is placed into shaking, however, samples could effectively be centrifuged rather than shaken. There are good reasons for choosing a shaking speed that keeps the solid phase in suspension and for maintaining conditions such as temperature constant throughout a study. In this way one can have confidence that the data have been obtained under the same conditions and therefore are comparable. If data from one study are to be compared with those from another study then consistency of methodologies and extraction conditions becomes important.
4.3 Presentation of data
Sequential extraction can generate a vast quantity of data. For each sample analysed using the four-step BCR procedure, five results are obtained for each element determined. Some procedures can have seven or eight steps. It is not always easy to present large datasets in a manner that is clear and easy to interpret. The use of bar charts is space-efficient and visually acceptable and so is widely adopted. Bar charts work particularly well if colour is used, but they are often less effective in black and white. Also, the large numbers needed in some studies can result in a reduction in size and a reduction in clarity of presentation (see, for example, Davidson et al.41).Unfortunately, the data in bar charts are in most cases presented as a percentage (with respect to either pseudototal values or the sum of the steps of the sequential extraction). This can be misleading unless the absolute levels of metals are also provided. For example, Fig. 2(a) suggests that element A has the greatest potential for mobilization and hence presents the greatest risk (assuming the three analytes have equal toxicities). However, when the same data are presented in terms of concentration [Fig. 2(b)], it becomes clear that all three elements have exactly the same concentrations in each of Steps 1, 2 and 3.
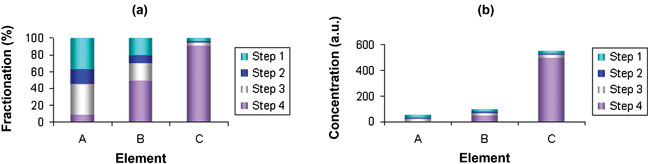 | ||
| Fig. 2 Hypothetical results of a BCR-type extraction, presented as (a) percentage fractionation patterns and (b) absolute concentrations. | ||
Potential confusion can easily be avoided by displaying the absolute amounts of metal in each fraction (see, for example, Ettler et al.142) or by presenting bar charts for both analyte concentrations and analyte fractionation patterns in the same figure, exemplified by Gonneea and Paytan143 in a recent study of barium in sediments using a five-step sequential extraction procedure.
Authors frequently base their discussion solely on the relative amounts of elements in each fraction and this can be misleading. Statements such as ‘percentage Pb bioaccessibilty was the lowest in the most contaminated soils’ could be misinterpreted because the most contaminated soils by definition have the highest concentrations so they could easily have the highest absolute amount of ‘bioaccessible lead’ even though the percentage is the lowest. Reimer et al.144 considered the high arsenic levels in crushed rocks to be unavailable because only 10% was easily extracted, yet low concentrations of arsenic in organic soils were ‘more available’ because 10–50% was extracted. It is the absolute amount, however, that is important. This was recognized by Abollino et al.145 when they justified presentation of relatively small extractable amounts of aluminium because ‘0.004% Al extracted into the exchangeable fraction corresponds to 3.1 mg kg–1 of the element’.
Some studies146–148 have used a ‘Risk Assessment Code’ to assess the environmental impact of contaminated river sediments. In this code the degree of risk is related to the relative amount of metals associated with the ‘carbonate’ and ‘exchangeable’ fractions. A value of <1% is considered to present no risk to the aquatic environment even though 1% of a large concentration could still be harmful. It is questionable whether a risk assessment should be based on the percentage of metal extracted in a particular fraction.
A similar caveat applies to the presentation of PTE mobility series based on the proportion (percentage) of each element released by the initial step or step(s) of a sequential extraction. Considering again the hypothetical sequential extraction presented in Fig. 2, a mobility series for elements A, B and C could be proposed based on Step 1 [A(36%) > B(20%) > C(4%)] or on the fractions associated with non-residual components [Σ(Steps 1–3): A(91%) > B(50%) > C(9%)]. Both are potentially misleading and fail to properly emphasize the fact that a very small proportion of a large amount of PTE could be much more significant than a high proportion of a small amount.
4.4 Interpretation of data
Once data have been acquired then they must be interpreted. The interpretation depends strongly on the context and aim of the study so that one metal pool could be considered as ‘bioavailable’ in studies of plant uptake yet ‘mobile’ or ‘labile’ in leaching studies. Surprisingly, many studies still make no attempt to interpret the data obtained within a clearly-defined context.Interpretation is not aided by the dependence on the operations used to obtain individual fractions. Not only is it necessary to understand the context of the sample but also the implications of adopting a particular procedure. It is important to understand the differences between the two BCR procedures so that appropriate conclusions can be drawn. It is important to understand that the Tessier and BCR procedures will not necessarily give the same results. For example, Mn is extracted from agricultural soils predominantly in the reducible fraction of the BCR procedure, but predominantly in the residual fraction of the Tessier scheme.28 It is therefore important to realize that comparison with data from outside a study should only be done with caution.
Consistent interpretation of the outputs of sequential extraction is further hampered by the large variations that exist in authors' definitions of ‘bioavailable’, ‘mobile’ and ‘labile’ pools. Bioavailability is a complex and evolving concept,149 but has recently been defined as the ‘degree to which chemicals present in the soil may be absorbed or metabolized by a human or ecological receptor or are available for interaction with biological systems’.150 Whilst it is entirely legitimate to use sequential extraction as a tool to deduce information on the potential bioavailability of PTEs, due emphasis must be placed on ‘potential’. A large number of factors (physical, chemical and physiological) affect whether an element will be incorporated into an organism: even those elements extracted early in a sequential extraction are not necessarily currently bioavailable and may never become so.
Similar comments apply to the designation of PTEs in particular fractions as ‘mobile’ or ‘labile’. Whether mobilization will actually occur depends on a large number of additional environmental factors, including the prevailing pH, redox conditions, and availability of solid and colloidal particulate phases for re-adsorption. Sequential extraction can thus only indicate the potential, rather than the actual, mobility of soil- and sediment-bound species.
Despite the fact that sequential extraction schemes are designed with increasing reagent activities as one proceeds through the steps, few authors interpret the data as reduced availability or mobility passing through the stages of the extraction.147,151 This is probably because knowing that one fraction is more available than another is not necessarily helpful and researchers prefer to try to put absolute qualities to their data.
The majority of papers define the ‘bioavailable/mobile pool’ as the most easily extracted fractions (corresponding to the first step of the BCR procedures and the first two steps of the Tessier procedure).152–185 However, a substantial number of papers define the ‘bioavailable/mobile pool’ as all fractions except the residual fraction (corresponding to all three steps of the BCR procedure or all four steps of the Tessier method).78,79,186–202 It goes without saying that these two definitions can result in quite different interpretations of data. There are also a number of intermediate definitions in which the reducible fraction203–210 or oxidisable fraction211–217 was considered to represent the ‘bioavailable/mobile’ pool, either together with the easily extractable pool or on their own. Distinction is also made in some studies between the easily reducible pool (more mobile) and less easily reducible pool (less mobile).218,219 In contrast the reducible220,221 or oxidisable222,223 fractions have been specifically described as immobile or unavailable.
There are, however, some subtle (and some less subtle) nuances to the meaning of ‘bioavailable’ or ‘mobile’ fractions. Whereas the ‘water soluble’ and ‘carbonate’ fractions have been defined simply as ‘mobile’155 or ‘bioavailable’,157 other descriptions of the ‘exchangeable’ fraction range from ‘mobilizable’,183 the fraction with ‘greatest potential for chemical remobilization’,181 the ‘form of high mobility and potential bioavailability’,179 ‘readily available’156 to ‘highly mobile and bioavailable’.153
Interpretation is clearly not straightforward and, to quote De Vries et al.,224 ‘despite numerous sequential extraction schemes, specific information on the availability of those pools in view of its relation with dissolved concentrations, readily available for plant uptake or leaching to groundwater is lacking’.
Improved understanding of the relationships between results of sequential extraction and measured biological uptake can be obtained by analyzing samples of biota in parallel with sequential extraction of the soils or sediments on which they live. Many studies of this type have appeared in recent years, focusing on a variety of organisms: microbes;40,225,226 plants (lettuce,227–230 alfalfa,231 wheat,226,232–235 maize,236 brassicas,132,182,237 trees,238 rice,239 peanut,240 basil,241 Swiss chard,241 sea rush,242 tall fescue,243 tea,244 corn,245 parsley,230 dill230 and onion230) and animals (mussels,246–249 rag worms,249 fish,250 spiders,251 earthworms44,174,252,253 and rats254). However, considerable further research is needed.255,256
There is contradictory evidence as to whether a correlation exists between extractable concentrations of metals and some observable biological effect such as plant uptake. Whereas no correlation was found between uptake by wheat,234 sorghum257 or plantain258 and extractable metal contents, good correlation has been found between plant uptake and the metal concentrations extracted in the first fraction of sequential extraction schemes.244,259–262 Up to 19% of the 1500 mg kg–1 lead found in polluted paddy fields was extracted in the first step of a sequential extraction procedure and corresponded to a high uptake of lead by rice plants.261 Removal of copper from contaminated soil by poly(amidoamine) dendrimers correlated well with the ‘exchangeable and carbonate-bound’ copper fractions.263 Uptake of metals by vegetables,230 corn264 and sugarcane265 all correlated with either the ‘exchangeable’ or ‘carbonate-bound’ fractions together with the ‘organic’ fraction which were therefore considered to be ‘bioavailable’. In contrast the uptake of copper and zinc from sludge-amended soils correlated with the metals extracted in the ‘reducible’ fraction.266 In pot trials, zinc uptake seemed to correlate with all extractable fractions.267
The contradiction in evidence is exemplified by two studies on the uptake of metals by plants following amendment of soil with sewage sludge. Mendoza et al.257 found that uptake by sorghum of metals did not correspond to the availability predicted by sequential extraction. The authors concluded that neither single nor sequential extractions provided a good prediction of metal bioavailability. Pedra et al.268 on the other hand concluded that both total and extractable metal concentrations gave adequate predictions of plant uptake.
Evidence from the use of direct bioaccessibility assays has also proved somewhat contradictory. Use of a microbiological biosensor for lead indicated that the first stage of the BCR procedure overestimated the ‘bioavailable’ fraction of lead and that a single extraction with CaCl2 gave a better correlation.269 Similarly, a bioluminescence assay gave a lower ‘bioavailable’ fraction of lead than the first stage (1 M sodium acetate) of a sequential extraction test.270 The bioluminescence assay gave a higher value for ‘bioavailable’ copper, however, than the oxidation step of the sequential extraction procedure. Use of a stable-isotope-dilution procedure revealed that there was no correspondence between the isotopically labile cadmium pool in soils, often taken as representing the bioavailable pool, and the first fraction of a sequential extraction procedure.271 This led the authors to state that ‘results suggest that conventional SEPs may be of limited utility for predicting bioavailability, for example, during ecological risk assessment’.
In contrast to these findings, a good correlation was found between ‘bioaccessible’ cadmium, as measured using a physiologically-based in vitro test, and water-soluble and ‘exchangeable’ cadmium.272 A reduction in the ‘bioavailability’ of lead, measured with an in vivo test using rats, when biosolids were incorporated into contaminated soils was reflected by a change in partitioning towards ‘less available’ phases as measured by sequential extraction.254 In a study on the toxicity to microorganisms of copper in soils, a correlation existed between IC50 (a measure of toxicity) and the exchangeable copper fraction but not with any of the other fractions.273 The bioavailability of mercury in sediments, measured using the assimilation efficiency in the clam, correlated well with mercury identified as bound to Fe/Mn oxide, amorphous organosulfur or the mineral lattice but not with mercury identified as bound to organocomplexes.274
In addition, or as an alternative, to bioassay, some authors have compared sequential extraction with the use of well-established, single extraction procedures, generally assumed to provide a reasonable estimate of plant-available PTEs in soil, e.g.diethylenetriamine pentaacetic acid (DTPA)52,182,254,275–277 and ethylenediamine tetraacetic acid (EDTA).276,278–281 Relationships between sequential extraction data and results of physiologically-based extraction tests designed to estimate PTE bioaccessibility in humans – following (usually) accidental intake of contaminated soil or sediment – are also beginning to be explored.133,226–228,269,272
5. Recent applications
Given the problems and uncertainties associated with sequential extraction analyses, one would be forgiven for wondering why anybody bothers using them. The fact that sequential extraction continues to see increased use suggests that researchers see the procedure as providing useful information. Examples of applications already cited in previous sections indicate that under certain conditions the information provided by sequential extractions correlates well with observable effects. An additional example is the fascinating find that high levels of ‘bioavailable’ copper in contaminated beach sediments, as measured by sequential extraction, correlated with low biodiversity.282 Is this direct evidence of a link between ‘bioavailable’ copper measured by sequential extraction and a toxic effect?Sequential extractions are relatively complicated, time-consuming and expensive and so should be only used when appropriate. They are not a very cost-effective method, for example, for predicting plant uptake of metals from contaminated soils. There is generally no advantage over the traditional use of single extractants such as EDTA28 or CaCl2.269 Similarly, single extractants have been found better than sequential extractions for the study of sediments.283 Even the use of multiple single extractants, essentially those used in the Tessier sequential scheme, has been proposed as a preferred option.284 This last proposal raised a number of issues such as double-accounting, however, which lie outside the scope of this review.
The lack of specificity is in itself not a major disadvantage in environmental studies. The chosen operations match conditions found within the environment, in particular the redox conditions, so it can be argued that it is more important to know the levels of metals susceptible to release under reducing or under oxidising conditions rather than to know the specific phases that are being attacked.
Taken together with other information, sequential extraction can provide information on natural processes within soils and sediments. Whereas soil organic matter was considered to play an important role in immobilizing lead in forest soils,43 the Fe–Mn oxides controlled metal mobilization in sediments contaminated by mining activity.285 The finding from isotope analysis that anthropogenic lead had penetrated no deeper than 10 cm in forest soils was consistent with the observation that lead below this depth was in a residual form.286 In environments dominated by limestone, carbonate species played a major role in the ‘self-purification’ of waters287 which was seasonal.288
Sequential extraction is widely used to assess the impact of human activity such as mining on the environment.45,47,289,290 Such studies are enhanced if a link can be demonstrated between metal fractionation and a biological affect such as intake by fish.250 These studies are most effective when comparing two different states. Changes can be observed in metal fractionation in the sediments of an estuary and those of its upper catchment in a mining area291 or between contaminated river sediments and mud.108 Seasonal changes in metal partitioning have been observed for sediments.292 The activity of indigenous sulfur-oxidising bacteria in contaminated sediments changed metal distributions with consequent remobilization of metals.293 Human activity can have an impact in a wide range of cases including the addition of copper to reservoirs as an algicide,294 the use in road construction of ashes from the incineration of municipal waste57 and the generation of urban air particulates.60 Sequential extraction has been used with other tests to investigate the effectiveness of leach tests which are widely used to assess release of metals from mineral processing waste.49
Classic examples of ‘before and after’ application of sequential extraction are the numerous studies on the clean-up of wastes and the remediation of contaminated sediments and soils. In all these studies, sequential extraction is used to determine metal partitioning both before and after the treatment thereby allowing an assessment of the effectiveness of the treatment to be made. Here is a strength of sequential extractions. Although it might not be possible to assign metals to specific soil phases, a general change in their reactivity can be identified and the consequences for their ‘bioavailability’ or ‘mobility’ assessed. There are two major approaches to the remediation of contaminated soils and sediments. One is the removal of metals through techniques such as acid washing,295 the use of chelating agents296–298 or other extractants,299 microbially-produced surfactants300,301 or flotation techniques.302 Sequential extraction can be used to evaluate the potential of electrodialytic remediation of harbour sediments, a technique which requires the metals to be in a mobile form.303 Similarly, the use of biodegradable chelating agents is only effective if the metals are present in mobile forms.296 The effectiveness of electrokinetic remediation methods was highly dependent on soil type and conditions.304,305 The observation304 that up to 76% copper could be removed from one soil but that copper was only re-distributed between fractions in another soil led to a caution against generalization of the remediation process.
The second approach to the remediation of soils is through the use of inorganic amendments to immobilize metals.306–308 Sequential extraction is used to confirm that metals are bound in less extractable forms after amendment of the soils. Relatively large amounts of a mixture of lime and fly ash reduced the leachability of metals, but if insufficient amendment was added the leachability of some metals could actually increase.309 Some procedures were only partially successful. Addition of 1% iron grit to soil, for example, successfully decreased the availability of arsenic and chromium and resultant uptake by plants but had no effect on copper uptake.310 In contrast, amendment of contaminated soil with a mixture of coal fly ash and peat reduced the leachability of copper and lead substantially.311 The most widely used inorganic amendment is the addition of phosphate to soils to immobilize metals.132,312–317 These studies consistently found that lead was converted into pyromorphite, a stable lead mineral, but the effectiveness has been reported to be greater for a soluble form than for the widely used rock phosphate312 and to be dependent on grain size.315 Sequential extraction was used to assess the effect on plant uptake of mine tailings with sewage sludge.318 Organic amendments such as humic acids319 also immobilized metals in contaminated soils. Heat treatment alone was sufficient to fix radionuclides in soils.320
Amendment of soils with organic wastes is widely seen as a beneficial means of utilizing these wastes, but there are clear implications in the possible introduction of harmful compounds including metals into soils and the wider ecosystem. Analysis of sewage sludges revealed that metal partitioning within the sludge was strongly influenced by the stabilization treatment used in production of the sludge.52,321 Most studies have, however, addressed the question of what happens to the metal distribution once the sludge is introduced into the soil. Although significantly increased amounts of extractable metals have been reported for sludge-amended soils322 or for soils irrigated with waste water323 and no change in zinc distribution has been observed,30 the general observation is that metal availability reduces and the metals become associated with residual phases in the soil.29,324–326 Whereas cadmium introduced into soil with chicken or pig manure was found predominantly in the unavailable residual fraction,327cadmium spike added to municipal waste composts was associated in the soil mainly with the relatively bioavailable fractions.328 In calcareous soils, most of the metals in sewage sludge became associated with the ‘carbonate’ and other relatively available fractions.329 Soils amended with paper mill sludges,31 tannery sludges33 and municipal solid waste compost32 have also been investigated. The thermal treatment given by forest fires reduced metal availability in sludge-amended forest soils.330
Composting of organic wastes is seen as a means of reducing metal availability prior to introducing the waste into soil. Composting of municipal solid waste changed metal partitioning with a shift to residual forms.54 Although some studies confirmed that composting of sewage sludge reduces the ‘availability’ of metals,51,55,56,331 this was not found consistently and increases in cadmium51 and lead50 ‘availability’ have been reported. Analysis of a range of different composts confirmed that composting altered the metal distribution within the waste but also that the changes in distribution depended on the source of the compost.53 The removal of heavy metals from sewage sludge prior to use as a fertilizer has been investigated using chelating agents,332 bacterial leaching333 or electrokinetic treatment.334–336 Liming of sewage sludge to make the sludge stable for storage had the effect of re-distributing copper in the oxidisable fraction to both the exchangeable and the residual fractions.337 Metals in incinerator fly ash could be made less reactive by amending the ash with clays.338 Cement-stabilized sewage sludge, intended for use as an artificial soil in earth works, had reduced levels of ‘available’ zinc but increased levels of ‘available’ copper.339
Studies on natural processes in soils help us to understand the action of plant exudates within the rhizosphere in increasing the bioavailability of trace elements.34,38,39,42,340,341 Such studies can also provide a better understanding of how hyperaccumulator plants mobilize and take up heavy metals and so can be used for phytoremediation.35,36,48,342,343 The effectiveness of phytoremediation can be improved by adding a chelating agent such as EDTA to the soil in order to increase the ‘bioavailability’ of the metals.243,344 The effectiveness of microbial recolonization of mine tailings has also been demonstrated.225 Other soil processes studied include the role of mycorrhiza in immobilizing metals and thereby protecting the host plants,37 the effect of earthworms on metal availability in contaminated soils44 and changes in metal partitioning resulting from increased humic acid levels in soils.345
Recent studies provide good examples of the wide range of environmental issues to which sequential extractions have been applied. Over time, zinc introduced into soil became less ‘available’, a so-called ‘aging’ process.346 Snowmelt runoff resulted in episodic releases of oxidation products from mine waste deposits347 whereas metals washed from roads into retention ponds were firmly bound in the sediments.348 A comparison of road de-icing compounds found that whereas the use of NaCl had no significant effect on cadmium ‘bioavailability’, the use of potassium formate reduced cadmium ‘bioavailabilty’ in soils significantly and was therefore the preferred option.349 Industrial discharges of chelating agents directly into the River Nile were considered to result in great remobilization of metals in the river sediments.350 A modern problem is the primitive processing of e-waste in China which has led to serious contamination of waters and sediments and could be impacting on both the health of local residents and the quality of the downstream environment.351
6. Emerging developments and trends
6.1 Ultrasound- and microwave-assisted extraction
A major disadvantage of sequential extraction is that it is time-consuming. For example, the BCR procedure involves three periods of overnight shaking. Together with aqua regiadigestion of the residue, and analysis of extracts and digests, this means that an entire week may be required to obtain results from a batch of samples – a fact hardly likely to encourage widespread use of the approach in busy, environmental monitoring laboratories. Also, whilst shaking can be carried out unattended, procedures such as oxidation and acid digestion are labour-intensive. There is thus considerable interest in developing new approaches that generate information similar to conventional sequential extraction but are faster to implement.Ultrasound is increasingly finding applications in analytical chemistry as a tool for rapid and efficient leaching of analytes from samples352 and various authors have attempted to develop ultrasound-assisted sequential extraction procedures. Perez-Cid et al.353 used the reagents recommended in the original BCR protocol and a sample of urban sewage sludge to develop the first such extraction, reported in 1998. Sonication conditions were optimized so that the amounts of metals extracted in the various steps matched, as far as possible, those obtained by conventional shaking. Ultrasound-assisted versions of the Tessier protocol, developed for both sewage sludge354 and river sediment,355 soon followed.
Various workers have since developed and applied ultrasound-assisted variants of both Tessier356,357 and BCR73,76,307,358–363extractions. However, uncertainties remain about whether these truly access the same phases as standard procedures, as demonstrated, for example, by difficulties experienced in obtaining similar fractionation patterns for matrix elements such as iron73,358 by conventional and ultrasonic extraction – even though results for trace elements were similar. Many extraction procedures have been developed on the basis of reference materials but have yet to be proved on real samples.73,360,362,364 Some have been optimized specifically with a view to application only to a particular sample type, e.g. sewage sludge.76,353,354 Whether such methods are transferable to other substrates remains somewhat questionable.358
Microwave-assisted sequential extraction procedures also exist. Again, most research has focused on well-established extraction schemes. Microwave-assisted protocols simulating the Tessier extraction have been developed based on lagoon sediment,365 sewage sludge,366 river sediment,367 and fly ash.368,369 Protocols based on the BCR extraction are reported for estuarine,360 marine370 and freshwater362 sediment, and also for fly ash.368 Work has also been conducted on pond sediment,371 vehicle exhaust particulates,371 calcareous soils372 and, uniquely, coal.373 Use of a set of microwave-assisted single extractions as a time-saving alternative to conventional sequential extraction has also been proposed.374,375
There is considerable merit in the development of these rapid approaches. However, the physical processes involved in ultrasound or microwave treatment are not the same as mechanical agitation. Also, considerable heating can occur in steps normally performed at room temperature. Owing to the operational nature of sequential extraction, the development of ultrasound- or microwave-assisted protocols that give similar performance to conventional shaking for all types of substrates seems unrealistic. More probable is the acceptance of standard protocols, perhaps using the same reagents as the Tessier or BCR procedures but not necessarily generating the same PTE fractionation patterns as conventional shaking, which can be used by laboratories to obtain harmonized data.
6.2 Dynamic extraction
An alternative means to achieve rapid results is to load the soil or sediment sample into a suitable container and perform the sequential extraction in continuous flow mode. Fritted centrifuge tubes,345,376–378 microcolumns379–382 and rotating coiled columns383–387 have been used for this purpose. Dedicated extraction cells388 have been developed including one combining continuous-flow operation and microwave irradiation.389Performing sequential extraction in dynamic mode overcomes potential problems associated with analyte re-adsorption during prolonged contact between extract and residual solids.390 The extractograms obtained provide information on element associations391–393 and, with use of suitable reagent flow rates, some systems can be coupled directly to FAAS,381ICP-OES383 or ICP-MS379,388 instruments. Also, the change from thermodynamic (i.e. equilibrium) to kinetic control of the leaching process has been claimed to represent more accurately environmental processes such as the percolation of rainwater through a soil profile. The reagents applied are usually similar to those used in conventional sequential extraction, although some authors have selected non-specific reagents, such as increasing concentrations of nitric acid.379
Workers at the British Geological Survey have developed and successfully applied a rapid extraction in which a series of non-specific extractants is drawn through the sample by centrifugation, and the resulting extractograms interpreted by chemometrics to obtain information on geochemical associations of PTEs.345,376–378 Dynamic sequential extraction has been recently reviewed by Miro and co-workers.68,394
6.3 Use of chemometrics
As in many other areas of analytical science, the past decade has seen a marked increase in the application of multivariate data analysis procedures as tools in the interpretation of results obtained by sequential extraction. Not only is it possible to assess relationships between (sometimes large numbers of) sampling sites and analytes, it is also feasible to include additional parameters, such as general soil or sediment characteristics, to gain a better overall insight into factors governing the environmental behaviour of PTEs.395,396 Most authors employ principal components analysis (PCA), but there are recent studies involving use of the Tucker N-way method397 and fuzzy clustering algorithms.398Chemometric processing of data obtained from sediment fractionation studies has helped inform decisions about land management,399 provided insight into the geochemical effects of urbanization,400 revealed associations between various groups of metals194,401–403 and provided information about anthropogenic sources of contaminants in lakes404 and river systems.192,223,405–408 Pollution source apportionment has also been achieved in street dusts.79,202 Sewage sludge has been studied to evaluate its suitability for use as an agricultural amendment.409
An early application of PCA to results obtained in the Tessier extraction of soil reference material SRM 2710 provided clear evidence of non-specificity.410 Later studies explored correlations between results of different extraction procedures.281 The combination of correlation analysis, PCA and hierarchical cluster analysis has been used to investigate relationships between metals recovered in the various steps of the BCR sequential extraction and plant uptake in an urban garden.411 Chemometric treatment has also been used to study the effects of land-use412 on soil metals and to identify soils requiring remediation on the basis of high concentrations of PTEs in easily extractable forms.145
6.4 Sequential extractions combined with stable isotope analysis
Although sequential extractions alone can provide useful information on the association of metals with different reactive phases in soils and sediments, combined with isotope analysis they become a powerful technique which can provide information on the origin of the metal and on soil processes. Whereas total concentration and bulk isotope analysis revealed no penetration of anthropogenic lead into soil profiles, the combination of sequential extraction and isotope analysis demonstrated this clearly.413 When combined with precise isotope analysis, use of this approach was able to distinguish between the lead in residual components from three nominally identical soils.414Because of concern arising from the widespread dispersion of lead throughout the environment, this approach has been applied principally to the study of lead which, fortuitously, has a variable isotopic composition in nature. Typical studies include those on estuarine sediments,415 freshwater sediments,416,417 agricultural soil,418 organic-rich soils,419 forest soils420 and contaminated soils.421 In forest soils422 and contaminated marine sediments,140 most anthropogenic lead was associated with ‘organic matter’ whereas, in lake sediments,423 anthropogenic lead was associated predominantly with the ‘oxide-hydroxide’ fraction.
A striking common finding in the studies using lead isotopic composition is that lead in the different extracted fractions, no matter the operations used, is isotopically distinct. Even though sequential extractions should not be considered as targeting specific phases, the results of these studies indicate that the operations are extracting distinct fractions. The unequal isotopic distribution of lead between different fractions was true even at depths in St Lawrence sediments where no industrial lead was to be expected.141
Although most studies exploiting the variable isotopic composition of strontium to investigate the provenance of strontium in soils and sediments have used single extractant tests, some use of sequential extraction combined with highly precise strontium isotope analysis has been made. Yokoo et al.424 demonstrated that an extraction scheme, designed to attack specific mineral phases, could be used to identify the provenance of both wet and dry deposition in loess and sand from China. A combination of strontium and neodymium isotopic compositions and rare-earth element concentrations in acid-insoluble phases proved most suitable for characterizing dry deposition. Xu and Marcantonio425 used sequential extraction combined with strontium isotope analysis to investigate the distribution of elements in suspended particulates in the Mississippi River.
The development in recent years of highly precise mass spectrometers for isotope analysis has revealed that other elements can have variable isotopic composition and so could be suitable for this type of study. Emmanuel et al.426 investigated the use of iron isotopes as a tool for quantifying iron cycling in soils and demonstrated that extracted fractions have distinct δ57Fe signatures which could be used to calculate the isotopic composition of mixing end members.
Although all the studies previously cited exploited natural variations in isotopic composition, it is also possible to spike samples with enriched stable isotopes in order to investigate the incorporation of the spikes into different soil fractions and to study the long-term trace element dynamics in soils. Ahnstrom and Parker271 monitored the incorporation of cadmium spike into soil fractions defined by a five-step procedure to measure the total labile pool (‘E-value’) and the labile cadmium in each extracted fraction. The fact that no correspondence was found between the isotopically labile pool, often taken to represent the bioavailable pool, and the first step of the extraction procedure led the authors to conclude that ‘conventional sequential extraction procedures may be of limited utility for predicting bioavailability, for example, during ecological risk assessments’.
6.5 Application to ‘non-traditional’ analytes
The vast majority of sequential extraction literature focused on a small suite of PTEs: cadmium, chromium, copper, iron, lead, manganese, nickel and zinc. However, the approach is now being used more widely, with standard extraction procedures being applied to ‘non-traditional’ elements such as arsenic, mercury, selenium and radionuclides. Success has been limited, however, because of the markedly different chemistries of these elements from the metals and it is common practice for dedicated schemes to be developed for these other elements.Applications of arsenic speciation very much mirror those of heavy metal speciation. Studies of the natural distribution of arsenic, in particular in areas of high natural concentrations, included those on sediments,434,435 peat436 and coal.437 The majority of studies has, however, been targeted at assessing the mobility and availability of arsenic in mine wastes,432,438–445 contaminated sediments431,446–450 and contaminated soils.451–453 A particular application is to the behaviour of arsenic in paddy fields454 and other soils455 irrigated with waters with high natural concentrations of arsenic and to sequestration of arsenic in rice plants.456 The mobility of organo-arsenic pesticides in soils is of concern457–460 as demonstrated by the study of Sarkar et al.461 in which it was shown that arsenic was less bioavailable in soils with high concentrations of amorphous oxides, in particular of aluminium and iron. The concentrations in the soluble fractions correlated well with results from a physiologically-based extraction test. Phytoremediation of contaminated soils462–464 and the use of iron to stabilize contaminated soils433 or to purify waters465 have also received attention. An extraction procedure designed for the study of soils was applied to the analysis of lichens because most arsenic was present in trapped soil particles.466 The study of Han et al.467 on the speciation of arsenic in poultry waste was notable for defining the fractions operationally and not as specific phases. Over 47% of arsenic in the wastes was water-soluble, but when applied to soils, 72% of arsenic was extracted in the residual phase.
The limitations of sequential extractions for mercury speciation have been highlighted. Kim et al.473 found that specific phases were dissolving in the ‘wrong’ fractions of their procedure (Bloom et al.469) and reported ‘inconsistencies in speciation results between different extraction protocols’ which ‘might not yield accurate information’. In a comparison of different extraction schemes, Sladek and Gustin474 found that identification of specific mercury phases could only be made with caution and that the schemes overestimated the release of mercury from mine wastes. They concluded that ‘these methods require more extensive evaluation before they can be considered as a predictive measure of in situ volatilization and removal viawater’. On the other hand, Fernandez-Martinez et al.,475 whilst acknowledging that it was not possible to identify individual species, found differences in availability between soils and considered that the method ‘provides detailed information about mobility in soils’.
Mercury speciation has been applied predominantly to mine wastes,473,474,476 contaminated soils475,477–483 and contaminated sediments.484–487 An investigation into the use of willow trees to stabilize mercury in contaminated land found that the root systems were effective in trapping ‘bioavailable’ mercury.488
Schemes for selenium speciation can be either relatively simple or relatively complex and range from a three-step procedure to assess selenium in agricultural soils493 to the seven-step procedure of Kulp and Pratt.490 Sequential extractions have been applied to the study of selenium in soils,492–495 contaminated sediments,496 mine tailings497 and coal-bearing rocks.498
Sequential extraction procedures have been applied to investigate uranium geochemistry in rivers,504fertilizer-amended soils505 and Moroccan black shale oil.506 Environmental contamination arising from uranium mining507–509 and the technological enhancement of natural radionuclides by phosphates mining510 and production,511–513 coal burning and pyrite roasting514 and other industrial processes515 have been investigated. Studies have indicated that relatively small proportions of the transuranic elements plutonium and americium occur in easily extractable forms in contaminated soil516–518 and sediment.519 Hence, the potential for migration of these, highly chemically- and radio-toxic elements, once released to the environment, is generally rather limited.
Fractionation data have clarified the role of microbes in the geochemical cycling of 99Tc520 and of fungi in the cycling of 137Cs;521,522 the importance of sediments as a sink for 99Tc523 and 137Cs524 (released from the Sellafield nuclear reprocessing facility, Cumbria, UK); and the factors affecting the migration and fate of 60Co,525 137Cs,526,527 239/240Pu527 and 90Sr526 in soil. The approach has been used in assessing the efficiency of a heating method for immobilization of radionuclides in contaminated soil320 and as part of laboratory-based studies of radionuclide migration in rock fractures528 relevant to the design of nuclear waste repositories.
Sequential extraction has also provided considerable insight into the physico-chemical behaviour of radionuclides released by the Chernobyl accident (April 1986).529 It is now widely accepted that the majority of radiocaesium is strongly bound to soil components,530–533 whereas 90Sr is found predominantly in easily extractable forms, except close to the reactor where there is a strong influence of deposited fuel particles.530,531,533 Similar findings have been reported for river sediments in the Chernobyl area534 and confirmed by column and batch leaching experiments.535
7. Concluding remarks
Sequential extraction is an important and widely applied tool for gaining information on potential mobility (hence, potential bioavailability and toxicity) of PTEs in the environment. Despite the limitations highlighted in this review, the usefulness of sequential extraction is evident from considerable insights it has provided over almost three decades into the environmental behaviour of PTEs. Applications of the approach continue to expand, and now encompass more elements and substrates than were probably ever envisaged by early workers in the field.In response to the title of this review, we consider sequential extraction to have a healthy future in the 21st century but that its continued usefulness, in particular for environmental monitoring, requires researchers to be aware of the limitations. Studies based on sequential extractions are more likely to be successful if certain conditions are met:
(a) A ‘standard’ sequential extraction scheme (e.g. the revised BCR procedure) should be used whenever possible because of the availability of reference materials and the possibility of direct comparison between studies. The experimental work should be adequately validated through use of mass balance and/or certified reference materials.
(b) When using such a ‘standard’ scheme it is critical that the specified procedure is adhered to strictly or, at the very least, any variations should be reported.
(c) There should if possible be some degree of comparison within a study – that is, comparing the difference in ‘before and after’ situations, spatial or temporal variability, the effect of some form of treatment or other changes within a system. Because of their operational nature and the difficulty in interpretation, sequential extraction procedures are not particularly suited for absolute studies – that is, identifying the distribution of metals between specific soil phases without reference to any other analyses.
(d) Data should be presented in terms of absolute concentrations instead of or in addition to percentage values
(e) Data should be interpreted according to the ‘operational speciation’ definition of Ure.11 For example, PTEs recovered in Step 2 of the BCR procedure should be described as associated with the ‘reducible fraction’, rather than as being ‘bound to iron/manganese oxyhydroxides’.
(f) It is important that sequential extractions are not applied uncritically and users should take cognizance of the scope and limitations of the approach. Phenomena such as non-specificity and re-adsorption can occur to widely variable extents, depending on the substrate and PTE studied.
(g) Care should be taken when drawing inferences concerning bioavailability based on sequential extraction results. Although, in general, it is reasonable to expect that PTEs liberated early in a sequential extraction have the potential for more immediate environmental impact than those found in residual fractions, relationships between the results of sequential extraction and observed bioavailability depend on the element, the substrate, the organism studied and the exposure route.
Studies into the fundamental mechanisms of sequential extraction procedures are to be encouraged as these provide us with an improved understanding of the metal speciation within the matrix and the solid phases attacked by the extractants. Our understanding of the relationship between extractability on the one hand and mobility and bioavailability on the other needs to be improved. The availability of a wider range of reference materials would help ensure the production of reliable data and ease the comparison of data produced by different studies.
Acknowledgements
J. R. B. acknowledges the financial support provided by SEERAD (Scottish Executive Environment and Rural Affairs Department).References
- D. Littlejohn, C. Davidson, J. Bacon and P. Quevauviller, J. Soils Sediments, 2006, 6, 62 Search PubMed.
- A. M. Ure and C. M. Davidson, Chemical Speciation in the Environment, Blackwell, Oxford, 2nd edn, 2002 Search PubMed.
- J. J. D'Amore, S. R. Al-Abed, K. G. Scheckel and J. A. Ryan, J. Environ. Qual., 2005, 34, 1707 CrossRef CAS.
- A. Manceau, M. A. Marcus, N. Tamura, O. Proux, N. Geoffroy and B. Lanson, Geochim. Cosmochim. Acta, 2004, 68, 2467 CrossRef CAS.
- M. P. Isaure, A. Laboudigue, A. Manceau, G. Sarret, C. Tiffreau, P. Trocellier, G. Lamble, J. L. Hazemann and D. Chateigner, Geochim. Cosmochim. Acta, 2002, 66, 1549 CrossRef CAS.
- J. S. Bang and D. Hesterberg, J. Environ. Qual., 2004, 33, 891 CAS.
- T. A. Kirpichtchikova, A. Manceau, L. Spadini, F. Panfili, M. A. Marcus and T. Jacquet, Geochim. Cosmochim. Acta, 2006, 70, 2163 CrossRef CAS.
- T. Karlsson, P. Persson and U. Skyllberg, Environ. Sci. Technol., 2005, 39, 3048 CrossRef CAS.
- E. Welter, W. Calmano, S. Mangold and L. Troger, Fresenius’ J. Anal. Chem., 1999, 364, 238 CrossRef CAS.
- J. D. Ostergren, G. E. Brown, G. A. Parks and T. N. Tingle, Environ. Sci. Technol., 1999, 33, 1627 CrossRef CAS.
- A. M. Ure, Microchim. Acta, 1991, 2, 49 CrossRef CAS.
- D. M. Templeton, F. Ariese, R. Cornelis, L. G. Danielsson, H. Muntau, H. P. Van Leeuwen and R. Lobinski, Pure Appl. Chem., 2000, 72, 1453 CrossRef CAS.
- J. Hlavay, T. Prohaska, M. Weisz, W. W. Wenzel and G. J. Stingeder, Pure Appl. Chem., 2004, 76, 415 CrossRef CAS.
- A. Tessier, P. G. C. Campbell and M. Bisson, Anal. Chem., 1979, 51, 844 CrossRef CAS.
- A. C. Chang, A. L. Page, J. E. Warneke and E. Grgurevic, J. Environ. Qual., 1984, 13, 33 CAS.
- P. O. Scokart, K. Meeusverdinne and R. Deborger, Int. J. Environ. Anal. Chem., 1987, 29, 305 CAS.
- P. Quevauviller, G. Rauret, H. Muntau, A. M. Ure, R. Rubio, J. F. López-Sánchez, H. D. Fiedler and B. Griepink, Fresenius’ J. Anal. Chem., 1994, 349, 808 CrossRef CAS.
- A. M. Ure, P. Quevauviller, H. Muntau and B. Griepink, Int. J. Environ. Anal. Chem., 1993, 51, 135 CrossRef CAS.
- C. M. Davidson, P. C. S. Ferreira and A. M. Ure, Fresenius’ J. Anal. Chem., 1999, 363, 446 CrossRef CAS.
- A. Sahuquillo, J. F. López-Sánchez, R. Rubio, G. Rauret, R. P. Thomas, C. M. Davidson and A. M. Ure, Anal. Chim. Acta, 1999, 382, 317 CrossRef CAS.
- G. Rauret, J. F. López-Sánchez, A. Sahuquillo, R. Rubio, C. Davidson, A. Ure and P. Quevauviller, J. Environ. Monit., 1999, 1, 57 RSC.
- H. D. Fiedler, J. F. López-Sánchez, R. Rubio, G. Rauret, P. Quevauviller, A. M. Ure and H. Muntau, Analyst, 1994, 119, 1109 RSC.
- P. Quevauviller, G. Rauret, J. F. López-Sánchez, R. Rubio, A. Ure and H. Muntau, Sci. Total Environ., 1997, 205, 223 CrossRef CAS.
- J. F. López-Sánchez, A. Sahuquillo, H. D. Fiedler, R. Rubio, G. Rauret, H. Muntau and P. Quevauviller, Analyst, 1998, 123, 1675 RSC.
- G. Rauret and J. F. López-Sánchez, Int. J. Environ. Anal. Chem., 2001, 79, 81 CrossRef CAS.
- M. Pueyo, G. Rauret, D. Lück, M. Yli-Halla, H. Muntau, P. Quevauviller and J. F. López-Sánchez, J. Environ. Monit., 2001, 3, 243 RSC.
- A. Mocko and W. Waclawek, Anal. Bioanal. Chem., 2004, 380, 813 CrossRef CAS.
- J. M. Alvarez, L. M. Lopez-Valdivia, J. Novillo, A. Obrador and M. I. Rico, Geoderma, 2006, 132, 450 CrossRef CAS.
- D. Chaudhuri, S. Tripathy, H. Veeresh, M. A. Powell and B. R. Hart, Environ. Geol., 2003, 45, 115 CrossRef CAS.
- M. S. Yoo and B. R. James, Soil Sci., 2003, 168, 686 CrossRef CAS.
- S. Z. Zhang, S. X. Wang, X. Q. Shan and H. Z. Mu, Biol. Fertil. Soils, 2004, 40, 237 CAS.
- P. Bhattacharyya, A. Chakraborty, K. Chakrabarti, S. Tripathy and M. A. Powell, Chemosphere, 2005, 60, 1481 CrossRef CAS.
- A. K. Gupta and S. Sinha, Chemosphere, 2006, 64, 161 CrossRef CAS.
- S. Tao, Y. J. Chen, F. L. Xu, J. Cao and B. G. Li, Environ. Pollut., 2003, 122, 447 CrossRef CAS.
- H. Al-Najar, R. Schulz and V. Romheld, Plant Soil, 2003, 249, 97 Search PubMed.
- J. Yanai, N. Mabuchi, N. Moritsuka and T. Kosaki, Soil Sci. Plant Nutr., 2004, 50, 423 Search PubMed.
- Y. Huang, S. Tao and Y. J. Chen, J. Environ. Sci. (China), 2005, 17, 276 Search PubMed.
- Y. Yu and Q. X. Zhou, Biol. Fertil. Soils, 2006, 42, 450 CrossRef CAS.
- S. H. Ru, J. P. Xing and D. C. Su, J. Plant Nutr., 2006, 29, 921 CrossRef CAS.
- Y. G. Yang, C. D. Campbell, L. Clark, C. M. Cameron and E. Paterson, Chemosphere, 2006, 63, 1942 CrossRef CAS.
- C. M. Davidson, G. J. Urquhart, F. Ajmone-Marsan, M. Biasioli, A. D. Duarte, E. Diaz-Barrientos, H. Grcman, L. Hossack, A. S. Hursthouse, L. Madrid, S. Rodrigues and M. Zupan, Anal. Chim. Acta, 2006, 565, 63 CrossRef CAS.
- S. Inaba and C. Takenaka, Water, Air, Soil Pollut., 2005, 162, 285 CrossRef CAS.
- P. Sipos, T. Nemeth and I. Mohai, Environ. Geochem. Health, 2005, 27, 1 Search PubMed.
- T. Lukkari, S. Teno, A. Vaisanen and J. Haimi, Soil Biol. Biochem., 2006, 38, 359 CAS.
- W. D. Brady, M. J. Eick, P. R. Grossl and P. V. Brady, Soil. Sediment Contam., 2003, 12, 541 Search PubMed.
- D. J. Robertson, K. G. Taylor and S. R. Hoon, Appl. Geochem., 2003, 18, 269 CrossRef CAS.
- M. S. V. Basilio, K. Friese, J. C. de Lena, H. A. Nalini and H. M. P. Roeser, Quim. Nova, 2005, 28, 822 CAS.
- P. S. Kidd and C. Monterroso, Sci. Total Environ., 2005, 336, 1 CrossRef CAS.
- S. R. Al-Abed, P. L. Hageman, G. Jegadeesan, N. Madhavan and D. Allen, Sci. Total Environ., 2006, 364, 14 CrossRef CAS.
- Y. X. Chen, Y. M. Hua, S. H. Zhang and G. M. Tian, J. Hazard. Mater., 2005, 123, 196 CrossRef CAS.
- D. Gao, G. D. Zheng, T. B. Chen, W. Luo, W. Gao, Y. A. Zhang and Y. X. Li, J. Environ. Sci. (China), 2005, 17, 957 Search PubMed.
- A. Fuentes, M. Llorens, J. Saez, A. Soler, M. I. Aguilar, J. F. Ortuno and V. F. Meseguer, Chemosphere, 2004, 54, 1039 CrossRef CAS.
- Y. Y. Liu, T. Imai, M. Ukita, M. Sekine and T. Higuchi, Environ. Technol., 2003, 24, 1517 CrossRef CAS.
- K. Szymanski, B. Janowska and R. Sidelko, Asian J. Chem., 2005, 17, 1646 CAS.
- G. D. Zheng, T. B. Chen, D. Gao and W. Luo, Water Sci. Technol., 2004, 50, 75 Search PubMed.
- J. W. C. Wong and A. Selvam, Chemosphere, 2006, 63, 980 CrossRef CAS.
- J. Kumpiene, A. Lagerkvist and C. Maurice, Soil. Sediment Contam., 2006, 15, 429 Search PubMed.
- S. Sukandar, K. Yasuda, M. Tanaka and I. Aoyama, Environ. Pollut., 2006, 144, 726 CrossRef CAS.
- F. Fujiwara, M. Dos Santos, J. Marrero, G. Polla, D. Gomez, L. Dawidowskia and P. Smichowski, J. Environ. Monit., 2006, 8, 913 RSC.
- E. Dabek-Zlotorzynska, M. Kelly, H. D. Chen and C. L. Chakrabarti, Chemosphere, 2005, 58, 1365 CrossRef CAS.
- G. Laforest and J. Duchesne, J. Hazard. Mater., 2006, 135, 156 CrossRef CAS.
- N. Kaewkhomdee, C. Kalambaheti, S. Predapitakkun, A. Siripinyanond and J. Shiowatana, Anal. Bioanal. Chem., 2006, 386, 363 CrossRef CAS.
- A. V. Filgueiras, I. Lavilla and C. Bendicho, J. Environ. Monit., 2002, 4, 823 RSC.
- S. D. Young, H. Zhang, A. M. Tye, A. Maxted, C. Thums and I. Thornton, Soil Use Manage., 2005, 21, 450 Search PubMed.
- A. Sahuquillo, A. Rigol and G. Rauret, TrAC, Trends Anal. Chem., 2003, 22, 152 CrossRef CAS.
- O. Abollino, A. Giacomino, M. Malandrino and E. Mentasti, Ann. Chim. (Rome, Italy), 2005, 95, 525 Search PubMed.
- P. Smichowski, G. Polla and D. Gomez, Anal. Bioanal. Chem., 2005, 381, 302 CrossRef CAS.
- M. Miro and E. H. Hansen, Microchim. Acta, 2006, 154, 3 CrossRef CAS.
- X. D. Li, B. J. Coles, M. H. Ramsey and I. Thornton, Analyst, 1995, 120, 1415 RSC.
- M. D. Ho and G. J. Evans, Anal. Commun., 1997, 34, 363 RSC.
- G. Rauret, J. F. López-Sánchez, A. Sahuquillo, E. Barahona, M. Lachica, A. M. Ure, C. M. Davidson, A. Gomez, D. Lück, J. Bacon, M. Yli-Halla, H. Muntau and P. Quevauviller, J. Environ. Monit., 2000, 2, 228 RSC.
- R. A. Sutherland and F. M. G. Tack, Anal. Chim. Acta, 2002, 454, 249 CrossRef CAS.
- E. Dabek-Zlotorzynska, M. Kelly, H. Chen and C. L. Chakrabarti, Anal. Chim. Acta, 2003, 498, 175 CrossRef CAS.
- K. F. Mossop and C. M. Davidson, Anal. Chim. Acta, 2003, 478, 111 CrossRef CAS.
- J. Kubova, V. Stresko, M. Bujdos, P. Matus and J. Medved, Anal. Bioanal. Chem., 2004, 379, 108 CAS.
- T. G. Kazi, M. K. Jamali, A. Siddiqui, G. H. Kazi, M. B. Arain and H. I. Affidi, Chemosphere, 2006, 63, 411 CrossRef CAS.
- P. Matus, J. Kubova, M. Bujdos and J. Medved, Anal. Chim. Acta, 2005, 540, 33 CrossRef CAS.
- B. L. Larner, A. J. Seen and A. T. Townsend, Anal. Chim. Acta, 2006, 556, 444 CrossRef CAS.
- S. Kartal, Z. Aydin and S. Tokalioglu, J. Hazard. Mater., 2006, 132, 80 CrossRef CAS.
- M. Jakubowska, W. Zembrzuski and Z. Lukaszewski, Talanta, 2006, 68, 1736 CrossRef CAS.
- G. E. M. Hall, G. Gauthier, J. C. Pelchat, P. Pelchat and J. E. Vaive, J. Anal. At. Spectrom., 1996, 11, 787 RSC.
- D. Gomez, M. Dos Santos, F. Fujiwara, G. Polla, J. Marrero, L. Dawidowski and P. Smichowski, Microchem. J., 2007, 85, 276 CrossRef CAS.
- P. M. V. Nirel and F. M. M. Morel, Water Res., 1990, 24, 1055 CrossRef CAS.
- M. Sulkowski and A. V. Hirner, Appl. Geochem., 2006, 21, 16 CrossRef CAS.
- E. Tipping, N. B. Hetherington, J. Hilton, D. W. Thompson, E. Bowles and J. Hamilton-Taylor, Anal. Chem., 1985, 57, 1944 CrossRef CAS.
- J. Dodd, D. J. Large, N. J. Fortey, A. E. Milodowski and S. Kemp, Environ. Geochem. Health, 2000, 22, 281 Search PubMed.
- W. Calmano, S. Mangold and E. Welter, Fresenius’ J. Anal. Chem., 2001, 371, 823 CrossRef CAS.
- C. Ianni, N. Ruggieri, P. Rivaro and R. Frache, Anal. Sci., 2001, 17, 1273 CAS.
- F. C. Dominguez, J. Rodriguez and C. D. Y. Pomes, Can. J. Anal. Sci. Spectrosc., 2002, 47, 60 CAS.
- K. G. Scheckel, C. A. Impellitteri, J. A. Ryan and T. McEvoy, Environ. Sci. Technol., 2003, 37, 1892 CrossRef CAS.
- B. Dold, J. Geochem. Explor., 2003, 80, 55 CrossRef CAS.
- X. Wan, W. Wang, T. M. Ye, Y. W. Guo and X. B. Gao, J. Hazard. Mater., 2006, 134, 197 CrossRef CAS.
- J. S. Manchanda, V. K. Nayyar and I. M. Chhibba, Chem. Speciation Bioavailability, 2006, 18, 27 Search PubMed.
- S. W. Poulton and D. E. Canfield, Chem. Geol., 2005, 214, 209 CrossRef CAS.
- A. Neaman, F. Mouele, F. Trolard and G. Bourrie, Appl. Geochem., 2004, 19, 973 CrossRef CAS.
- C. Parat, J. Leveque, S. Dousset, R. Chaussod and F. Andreux, Anal. Bioanal. Chem., 2003, 376, 243 CAS.
- S. Tokalioglu, S. Kartal and G. Birol, J. Environ. Monit., 2003, 5, 468 RSC.
- P. Tlustos, J. Szakova, A. Starkova and D. Pavlikova, Cent. Eur. J. Chem., 2005, 3, 830 Search PubMed.
- C. Hanahan, Environ. Geol., 2004, 45, 864 CrossRef CAS.
- J. Lacal, M. P. da Silva, R. Garcia, M. T. Sevilla, J. R. Procopio and L. Hernandez, Environ. Pollut., 2003, 124, 291 CrossRef CAS.
- E. Peltier, A. L. Dahl and J. F. Gaillard, Environ. Sci. Technol., 2005, 39, 311 CrossRef CAS.
- E. D. Burton, I. R. Phillips and D. W. Hawker, Soil. Sediment Contam., 2006, 15, 253 Search PubMed.
- D. Dermatas, G. Shen, M. Chrysochoou, D. G. Grubb, N. Menounou and P. Dutko, J. Hazard. Mater., 2006, 136, 34 CrossRef CAS.
- E. Doelsch, B. Deroche and V. Van De Kerchove, Chemosphere, 2006, 65, 286 CrossRef CAS.
- J. Kalembkiewicz and E. Soco, Pol. J. Environ. Stud., 2005, 14, 593 Search PubMed.
- E. Fernandez, R. Jimenez, A. M. Lallena and J. Aguilar, Environ. Pollut., 2004, 131, 355 CrossRef CAS.
- K. A. Kryc, R. W. Murray and D. W. Murray, Anal. Chim. Acta, 2003, 487, 117 CrossRef CAS.
- S. R. G. Barreto, J. Nozaki, E. De Oliveira, V. F. Do Nascimento, P. Henrique, A. Aragao, I. S. Scarminio and W. J. Barreto, Talanta, 2004, 64, 345 CrossRef.
- L. Leleyter and F. Baraud, C. R. Geosci., 2005, 337, 571 Search PubMed.
- G. E. M. Hall, J. E. Vaive, P. Pelchat, G. F. Bonham-Carter, D. A. Kliza-Petelle and K. Telmer, Geochem.: Explor., Environ., Anal., 2006, 6, 163 CrossRef CAS.
- Y. Z. Lu, D. M. Dong, Y. Fu, X. E. Shen, M. Yuan and W. T. Sun, Chem. J. Chin. Univ., 2006, 27, 449 Search PubMed.
- W. Baeyens, F. Monteny, M. Leermakers and S. Bouillon, Anal. Bioanal. Chem., 2003, 376, 890 CAS.
- T. Hjorth, Anal. Chim. Acta, 2004, 526, 95 CrossRef CAS.
- K. Zehl and J. W. Einax, J. Soils Sediments, 2005, 5, 164 Search PubMed.
- R. D. Guy, C. L. Chakrabarti and D. C. McBain, Water Res., 1978, 12, 21 CrossRef CAS.
- C. Kheboian and C. F. Bauer, Anal. Chem., 1987, 59, 1417 CrossRef.
- P. P. Coetzee, K. Gouws, S. Pluddemann, M. Yacoby, S. Howell and L. Dendrijver, Water SA, 1995, 21, 51 Search PubMed.
- M. Raksasataya, A. G. Langdon and N. D. Kim, Anal. Chim. Acta, 1996, 332, 1 CrossRef CAS.
- X. Q. Shan and C. Bin, Anal. Chem., 1993, 65, 802 CrossRef.
- I. M. C. Lo and X. Y. Yang, Waste Manage. (Amsterdam, Neth.), 1998, 18, 1 Search PubMed.
- C. Whalley and A. Grant, Anal. Chim. Acta, 1994, 291, 287 CrossRef CAS.
- J. L. G. Ariza, I. Giraldez, D. Sanchez-Rodas and E. Morales, Talanta, 2000, 52, 545 CrossRef CAS.
- N. Belzile, P. Lecomte and A. Tessier, Environ. Sci. Technol., 1989, 23, 1015.
- J. L. Gomez-Ariza, I. Giraldez, D. Sanchez-Rodas and E. Morales, Anal. Chim. Acta, 1999, 399, 295 CrossRef CAS.
- M. D. Ho and G. J. Evans, Environ. Sci. Technol., 2000, 34, 1030 CrossRef CAS.
- E. A. Gilmore, G. J. Evans and M. D. Ho, Anal. Chim. Acta, 2001, 439, 139 CrossRef CAS.
- A. Bermond, Anal. Chim. Acta, 2001, 445, 79 CrossRef CAS.
- Z. S. Ahnstrom and D. R. Parker, Soil Sci. Soc. Am. J., 1999, 63, 1650 CAS.
- G. Rauret, R. Rubio, J. F. López-Sánchez and E. Casassas, Int. J. Environ. Anal. Chem., 1989, 35, 89 CAS.
- J. T. van Elteren and B. Budic, Anal. Chim. Acta, 2004, 514, 137.
- O. R. La, C. M. Barra, N. Sobrinho, N. Mazur and A. C. X. Velloso, Quim. Nova, 2003, 26, 323 CAS.
- Y. G. Zhu, S. B. Chen and J. C. Yang, Environ. Int., 2004, 30, 351 CrossRef CAS.
- K. G. Scheckel, J. A. Ryan, D. Allen and N. V. Lescano, Sci. Total Environ., 2005, 350, 261 CrossRef CAS.
- J. M. Kaste, A. J. Friedland and E. K. Miller, Water, Air, Soil Pollut., 2005, 167, 139 CrossRef CAS.
- C. M. Davidson, A. S. Hursthouse, D. M. Tognarelli, A. M. Ure and G. J. Urquhart, Anal. Chim. Acta, 2004, 508, 193 CrossRef CAS.
- Q. Tu, X. Q. Shan and Z. M. Ni, Sci. Total Environ., 1994, 151, 159 CrossRef.
- R. A. Sutherland and F. M. G. Tack, Adv. Environ. Res., 2003, 8, 37 Search PubMed.
- J. R. Bacon, P. Cooper and I. J. Hewitt, Chin. J. Geochem., 2006, 25, 202 Search PubMed.
- P. E. Jensen, L. M. Ottosen and A. J. Pedersen, Water, Air, Soil Pollut., 2006, 170, 359 CrossRef CAS.
- M. Kersten, C. D. Garbe-Schönberg, S. Thomsen, C. Anagnostou and A. Sioulas, Environ. Sci. Technol., 1997, 31, 1295 CrossRef CAS.
- C. Gobeil, W. K. Johnson, R. W. Macdonald and C. S. Wong, Environ. Sci. Technol., 1995, 29, 193 CAS.
- V. Ettler, M. Mihaljevic, O. Sebek, M. Molek, T. Grygar and J. Zeman, Environ. Pollut., 2006, 142, 409 CrossRef CAS.
- M. E. Gonneea and A. Paytan, Mar. Chem., 2006, 100, 124 CrossRef CAS.
- K. J. Reimer, C. A. Ollson and I. Koch, Biogeochem. Environ. Important Trace Elem., 2003, 835, 166 Search PubMed.
- O. Abollino, A. Giacomino, M. Malandrino, E. Mentasti, M. Aceto and R. Barberis, Water, Air, Soil Pollut., 2006, 173, 315 CrossRef CAS.
- C. K. Jain, Water Res., 2004, 38, 569 CrossRef CAS.
- E. Martley, B. Gulson, H. Louie, M. Wu and P. Di, Geochem.: Explor., Environ., Anal., 2004, 4, 171 CrossRef CAS.
- H. Ghrefat and N. Yusuf, Chemosphere, 2006, 65, 2114 CrossRef CAS.
- N. Calace, B. M. Petronio and M. Pietroletti, Ann. Chim. (Rome, Italy), 2006, 96, 131 Search PubMed.
- Report ISO 11074:2005, Soil quality – vocabulary, International Standards Organization, Geneva, Switzerland, 2005 Search PubMed.
- A. D. K. Banerjee, Environ. Pollut., 2003, 123, 95 CrossRef CAS.
- C. Aydinalp and S. Marinova, Pol. J. Environ. Stud., 2003, 12, 629 Search PubMed.
- G. Bird, P. A. Brewer, M. G. Macklin, D. Balteanu, B. Driga, M. Serban and S. Zaharia, Appl. Geochem., 2003, 18, 1583 CrossRef CAS.
- M. Imperato, P. Adamo, D. Naimo, M. Arienzo, D. Stanzione and P. Violante, Environ. Pollut., 2003, 124, 247 CrossRef CAS.
- X. D. Cao, L. Q. Ma, M. Chen, D. W. Hardison and W. G. Harris, Sci. Total Environ., 2003, 307, 179 CrossRef CAS.
- A. Navas and H. Lindhorfer, Environ. Int., 2003, 29, 61 CrossRef CAS.
- P. Kotoky, B. J. Bora, N. K. Baruah, J. Baruah and G. C. Borah, Chem. Speciation Bioavailability, 2003, 15, 115 Search PubMed.
- P. Janos, L. Herzogova, J. Rejnek and J. Hodslavska, Talanta, 2004, 62, 497 CrossRef CAS.
- G. Renella, P. Adamo, M. R. Bianco, L. Landi, P. Violante and P. Nannipieri, Eur. J. Soil Sci., 2004, 55, 123 CrossRef CAS.
- I. Ahumada, P. Escudero, M. A. Carrasco, G. Castillo, L. Ascar and E. Fuentes, J. Environ. Monit., 2004, 6, 327 RSC.
- A. Guevara-Riba, A. Sahuquillo, R. Rubio and G. Rauret, Sci. Total Environ., 2004, 321, 241 CrossRef CAS.
- K. Elass, A. Laachach and M. Azzi, Ann. Chim. (Rome, Italy), 2004, 94, 325 Search PubMed.
- F. Liu, H. G. Liu, Q. F. Yu and Y. F. Nie, J. Environ. Sci. (China), 2004, 16, 885 Search PubMed.
- E. Margui, V. Salvado, I. Queralt and M. Hidalgo, Anal. Chim. Acta, 2004, 524, 151 CrossRef CAS.
- S. Yu, Z. L. He, C. Y. Huang, G. C. Chen and D. V. Calvert, Geoderma, 2004, 123, 163 CrossRef CAS.
- Q. J. Song and G. M. Greenway, J. Environ. Monit., 2004, 6, 31 RSC.
- A. Giacalone, A. Gianguzza, S. Orecchio, D. Piazzese, G. Dongarra, S. Sciarrino and D. Varrica, Chem. Speciation Bioavailability, 2005, 17, 83 Search PubMed.
- S. Esakku, A. Selvam, K. Joseph and K. Palanivelu, Chem. Speciation Bioavailability, 2005, 17, 95 Search PubMed.
- F. Liu, J. G. Liu, Q. F. Yu, Y. Y. Jin and Y. F. Nie, J. Environ. Sci. Health, Part A, 2005, 40, 1975 Search PubMed.
- J. Virkutyte, E. van Hullebusch, M. Sillanpaa and P. Lens, Environ. Pollut., 2005, 138, 517 CAS.
- D. Wiechula, K. Loska and I. Korus, Water, Air, Soil Pollut., 2005, 164, 315 CrossRef CAS.
- P. K. Lee, Y. H. Yu, S. T. Yun and B. Mayer, Chemosphere, 2005, 60, 672 CrossRef CAS.
- E. Remon, J. L. Bouchardon, B. Cornier, B. Guy, J. C. Leclerc and O. Faure, Environ. Pollut., 2005, 137, 316 CrossRef CAS.
- T. Becquer, J. Dai, U. Quantin and P. Lavelle, Soil Biol. Biochem., 2005, 37, 1564 CrossRef CAS.
- A. S. Joksic, S. A. Katz, M. Horvat and R. Milacic, Water, Air, Soil Pollut., 2005, 162, 265 CrossRef CAS.
- S. Amir, M. Hafidi, G. Merlina and J. C. Revel, Chemosphere, 2005, 59, 801 CrossRef CAS.
- A. Navas and H. Lindhorfer, Soil. Sediment Contam., 2005, 14, 249 Search PubMed.
- G. Glosinska, T. Sobczynski, L. Boszke, K. Bierla and J. Siepak, Pol. J. Environ. Stud., 2005, 14, 305 Search PubMed.
- C. Wang, X. Hu, M. L. Chen and Y. H. Wu, J. Hazard. Mater., 2005, 119, 245 CrossRef CAS.
- K. M. Banat, F. M. Howari and A. A. Al-Hamad, Environ. Res., 2005, 97, 258 CrossRef CAS.
- G. Bird, P. A. Brewer, M. G. Macklin, M. Serban, D. Balteanu and B. Driga, J. Geochem. Explor., 2005, 86, 26 CrossRef CAS.
- Z. Y. Hseu, Chemosphere, 2006, 63, 762 CrossRef CAS.
- S. Audry, G. Blanc, J. Schafer, G. Chaillou and S. Robert, Geochim. Cosmochim. Acta, 2006, 70, 2264 CrossRef CAS.
- Q. J. Song and G. M. Greenway, Int. J. Environ. Anal. Chem., 2006, 86, 359 CrossRef CAS.
- I. Walter, F. Martinez and V. Cala, Environ. Pollut., 2006, 139, 507 CrossRef CAS.
- V. Saenz, J. Blasco and A. Gomez-Parra, Environ. Toxicol. Chem., 2003, 22, 2833 CrossRef CAS.
- J. Ciba, M. Zolotajkin, J. Kluczka, K. Loska and J. Cebula, Waste Manage. (Amsterdam, Neth.), 2003, 23, 897 Search PubMed.
- S. Lee, J. W. Moon and H. S. Moon, Environ. Geochem. Health, 2003, 25, 433 Search PubMed.
- M. Yaman and S. Bakirdere, Microchim. Acta, 2003, 141, 47 CrossRef CAS.
- C. G. Yuan, J. B. Shi, B. He, J. F. Liu, L. N. Liang and G. B. Jiang, Environ. Int., 2004, 30, 769 CrossRef CAS.
- M. C. Z. Moturi, M. Rawat and V. Subramanian, Environ. Monit. Assess., 2004, 95, 183 CrossRef CAS.
- A. V. Filgueiras, I. Lavilla and C. Bendicho, Sci. Total Environ., 2004, 330, 115 CrossRef CAS.
- P. Adamo, M. Arienzo, M. Imperato, D. Naimo, G. Nardi and D. Stanzione, Chemosphere, 2005, 61, 800 CrossRef CAS.
- D. Relic, D. Dordevic, A. Popovic and T. Blagojevic, Environ. Int., 2005, 31, 661 CrossRef CAS.
- M. A. Okbah, M. A. Shata and M. A. Shridah, Chem. Ecol., 2005, 21, 23 Search PubMed.
- A. Bielicka, I. Bojanowska and A. Wisniewski, Pol. J. Environ. Stud., 2005, 14, 145 Search PubMed.
- B. Walna, J. Siepak, S. Drzymala and T. Sobczynski, Pol. J. Environ. Stud., 2005, 14, 243 Search PubMed.
- S. E. Lee, J. S. Lee and H. T. Chon, in On the Convergence of Bio-, Information-, Environmental-, Energy-, Space- and Nano-Technologies, Pts 1 and 2, Trans Tech Publications Inc., Zurich, Switzerland, 2005, vol. 277–279, p. 438 Search PubMed.
- G. Garcia, A. L. Zanuzzi and A. Faz, Biodegradation, 2005, 16, 187 CrossRef CAS.
- J. Sysalova and J. Szakova, Environ. Res., 2006, 101, 287 CrossRef CAS.
- R. Fernandez-Martinez, J. Loredo, A. Ordonez and M. I. Rucandio, Environ. Pollut., 2006, 142, 217 CrossRef CAS.
- E. Tokalioglu and S. Kartal, Atmos. Environ., 2006, 40, 2797 CrossRef.
- M. Kaasalainen and M. Yli-Halla, Environ. Pollut., 2003, 126, 225 CrossRef CAS.
- M. Ranville, D. Rough and A. R. Flegal, Appl. Geochem., 2004, 19, 803 CrossRef CAS.
- C. X. Yang, Y. H. Chen, P. Peng, C. Li, X. Y. Chang and C. S. Xie, Sci. Total Environ., 2005, 341, 159 CrossRef CAS.
- J. S. Dube and R. Galvez-Cloutier, J. Environ. Eng. Sci., 2005, 4, 399 Search PubMed.
- F. Reith, D. C. McPhail and A. G. Christy, J. Geochem. Explor., 2005, 85, 81 CrossRef CAS.
- A. S. Knox, D. L. Dunn, M. H. Paller, E. A. Nelson, W. L. Specht and J. C. Seaman, Eng. Life Sci., 2006, 6, 31 CrossRef CAS.
- M. L. K. Rodrigues and M. L. L. Formoso, Water, Air, Soil Pollut., 2006, 169, 167 CrossRef CAS.
- E. Alonso, P. Villar, A. Santos and I. Aparicio, Waste Manage. (Amsterdam, Neth.), 2006, 26, 1270 Search PubMed.
- J. Morillo, J. Usero and I. Gracia, Chemosphere, 2004, 55, 431 CrossRef CAS.
- C. Durand, V. Ruban and A. Ambles, Environ. Technol., 2004, 25, 881 CAS.
- E. V. Hullebusch, F. Auvray, F. Bordas, V. Deluchat, P. M. Chazal and M. Baudu, Environ. Technol., 2003, 24, 787 CrossRef CAS.
- A. G. Sowder, P. M. Bertsch and P. J. Morris, J. Environ. Qual., 2003, 32, 885 CAS.
- S. Olivares-Rieumont, D. de la Rosa, L. Lima, D. W. Graham, K. D'Alessandro, J. Borroto, F. Martinez and J. Sanchez, Water Res., 2005, 39, 3945 CrossRef CAS.
- S. X. Wang, S. Z. Zhang, X. Q. Shan and H. Z. Mu, Sci. China, Ser. C, 2005, 48, 142 Search PubMed.
- C. Caplat, H. Texier, D. Barillier and C. Lelievre, Mar. Pollut. Bull., 2005, 50, 504 CrossRef CAS.
- G. P. Wang, J. S. Liu and J. Tang, Wetlands, 2004, 24, 608 Search PubMed.
- A. van der Veen, C. Ahlers, D. W. Zachmann and K. Friese, Acta Hydrochim. Hydrobiol., 2006, 34, 214 CrossRef CAS.
- Y. Li, J. J. Chen, X. L. Wang, D. M. Dong and S. H. Guo, Chem. J. Chin. Univ., 2006, 27, 627 Search PubMed.
- N. K. Erzen and J. Stupar, Acta Chim. Slov., 2003, 50, 67 CAS.
- J. W. Tang, G. R. Whittecar, K. H. Johannesson and W. L. Daniels, Geochem. Trans., 2004, 5, 49 Search PubMed.
- W. X. Liu, X. D. Li, Z. G. Shen, D. C. Wang, O. W. H. Wai and Y. S. Li, Environ. Pollut., 2003, 121, 377 CrossRef CAS.
- W. De Vries, J. Curlik, A. Muranyi, B. Alloway and B. J. Groenenberg, Ekologia (Bratislava, Slovakia), 2005, 24, 152 Search PubMed.
- J. V. Garcia-Meza, A. Carrillo-Chavez and O. Morton-Bermea, Environ. Geol., 2006, 49, 437 CAS.
- G. Siebielec, T. Stuczynski and R. Korzeniowska-Puculek, Pol. J. Environ. Stud., 2006, 15, 121 Search PubMed.
- N. Basta and R. Gradwohl, J. Soil Contam., 2000, 9, 149 Search PubMed.
- N. T. Basta, R. Gradwohl, K. L. Snethen and J. L. Schroder, J. Environ. Qual., 2001, 30, 1222 CAS.
- P. Castaldi, G. Garau and P. Melis, Fresenius Environ. Bull., 2004, 13, 1322 CAS.
- S. Tokalioglu, S. Kartal and A. Gultekin, Int. J. Environ. Anal. Chem., 2006, 86, 417 CrossRef CAS.
- X. D. Cao, X. R. Wang and G. W. Zhao, Chemosphere, 2000, 40, 23 CrossRef CAS.
- G. S. R. Krishnamurti and R. Naidu, Aust. J. Soil Res., 2000, 38, 991 Search PubMed.
- Z. W. Wang, X. Q. Shan and S. Z. Zhang, Chemosphere, 2002, 46, 1163 CrossRef CAS.
- Z. W. Wang, X. Q. Shan and S. Z. Zhang, Commun. Soil Sci. Plant Anal., 2003, 34, 1573 CrossRef CAS.
- H. R. Ma, X. R. Wang and C. B. Zhang, Chem. Speciation Bioavailability, 2003, 15, 15 Search PubMed.
- W. S. Wang, X. Q. Shan, B. Wen and S. Z. Zhang, Chemosphere, 2003, 53, 523 CrossRef CAS.
- P. Parkpian, S. T. Leong, P. Laortanakul and N. T. K. Phuong, J. Environ. Sci. Health, Part A, 2002, 37, 1099 Search PubMed.
- T. Punshon, K. F. Gaines, P. M. Bertsch and J. Burger, Environ. Toxicol. Chem., 2003, 22, 1146 CAS.
- Y. M. Luo, X. J. Jiang, L. H. Wu, J. Song, S. C. Wu, R. H. Lu and P. Christie, Geoderma, 2003, 115, 113 CrossRef CAS.
- D. Chaudhuri, S. Tripathy, H. Veeresh, M. A. Powell and B. R. Hart, Environ. Geol., 2003, 44, 419 CrossRef CAS.
- V. D. Zheljazkov and P. R. Warman, J. Environ. Qual., 2004, 33, 542 CAS.
- C. M. R. Almeida, A. P. Mucha and M. Vasconcelos, Environ. Sci. Technol., 2004, 38, 3112 CrossRef CAS.
- Y. X. Chen, J. Y. Shi, W. D. Zhang, Q. Lin and G. M. Tian, Plant Soil, 2004, 261, 117 Search PubMed.
- S. Tokalioglu and S. Kartal, Instrum. Sci. Technol., 2004, 32, 387 CrossRef CAS.
- H. F. Li, Q. R. Wang, Y. S. Cui, Y. T. Dong and P. Christie, Sci. Total Environ., 2005, 339, 179 CrossRef CAS.
- A. Jamil, K. Lajtha, S. Radan, G. Ruzsa, S. Cristofor and C. Postolache, Hydrobiology, 1999, 392, 143 Search PubMed.
- J. Pempkowiak, A. Sikora and E. Biernacka, Chemosphere, 1999, 39, 313 CrossRef CAS.
- C. K. Yap, A. Ismail, S. G. Tan and H. Omar, Environ. Int., 2002, 28, 117 CrossRef CAS.
- A. S. Hursthouse, J. M. Matthews, J. E. Figures, P. Iqbal-Zahid, I. M. Davies and D. H. Vaughan, Environ. Geochem. Health, 2003, 25, 171 Search PubMed.
- K. C. Sekhar, N. S. Chary, C. T. Kamala, D. S. S. Raj and A. S. Rao, Environ. Int., 2004, 29, 1001 CrossRef CAS.
- G. Du Laing, N. Bogaert, F. M. G. Tack, M. G. Verloo and F. Hendrickx, Sci. Total Environ., 2002, 289, 71 CrossRef CAS.
- C. J. Langdon, T. G. Piearce, A. A. Meharg and K. T. Semple, Environ. Toxicol. Chem., 2001, 20, 2336 CrossRef CAS.
- G. Maddocks, A. Reichelt-Brushett, D. McConchie and J. Vangronsveld, Environ. Toxicol. Chem., 2005, 24, 554 CrossRef CAS.
- S. Brown, R. L. Chaney, J. G. Hallfrisch and Q. Xue, J. Environ. Qual., 2003, 32, 100 CAS.
- D. R. Leece, G. K. C. Low, M. S. Warne, T. M. Manning, J. C. Chapman and K. Koop, Commun. Soil Sci. Plant Anal., 2000, 31, 2185 CrossRef CAS.
- G. S. R. Krishnamurti and R. Naidu, Environ. Sci. Technol., 2002, 36, 2645 CrossRef CAS.
- J. Mendoza, T. Garrido, G. Castillo and N. San Martin, Chemosphere, 2006, 65, 2304 CrossRef CAS.
- D. Lestan, H. Grcman, M. Zupan and N. Bacac, Soil. Sediment Contam., 2003, 12, 507 Search PubMed.
- M. Pueyo, J. Sastre, E. Hernandez, M. Vidal, J. F. López-Sánchez and G. Rauret, J. Environ. Qual., 2003, 32, 2054 CAS.
- J. M. Alvarez and M. I. Rico, J. Agric. Food Chem., 2003, 51, 5760 CrossRef CAS.
- Q. W. Yang, W. S. Shu, J. W. Qiu, H. B. Wang and C. Y. Lan, Environ. Int., 2004, 30, 883 CrossRef CAS.
- A. Obrador, J. Novillo and J. M. Alvarez, Soil Sci. Soc. Am. J., 2003, 67, 564 CAS.
- Y. H. Xu and D. Y. Zhao, Environ. Sci. Technol., 2005, 39, 2369 CrossRef CAS.
- M. R. Borges and E. L. M. Coutinho, Rev. Bras. Cienc. Solo, 2004, 28, 543 Search PubMed.
- E. I. Bertoncini, M. E. Mattiazzo and R. Rossetto, J. Plant Nutr., 2004, 27, 1243 CrossRef CAS.
- P. Bhattacharyya, A. Chakraborty, K. Chakrabarti, S. Tripathy and M. A. Powell, Environ. Geol., 2006, 49, 1064 CrossRef CAS.
- E. M. Andre, M. C. P. Cruz, M. E. Ferreira and L. A. S. Palma, Rev. Bras. Cienc. Solo, 2003, 27, 451 Search PubMed.
- F. Pedra, H. Domingues, A. B. Ribeiro, A. Polo and O. Monteiro, Environ. Technol., 2006, 27, 1357 CrossRef CAS.
- W. Geebelen, D. C. Adriano, D. van der Lelie, M. Mench, R. Carleer, H. Clijsters and J. Vangronsveld, Plant Soil, 2003, 249, 217 Search PubMed.
- P. Peltola, A. Ivask, M. Astrom and M. Virta, Sci. Total Environ., 2005, 350, 194 CrossRef CAS.
- Z. A. S. Ahnstrom and D. R. Parker, Environ. Sci. Technol., 2001, 35, 121 CrossRef CAS.
- X. Y. Tang, Y. G. Zhu, Y. S. Cui, J. Duan and L. Tang, Environ. Int., 2006, 32, 682 CrossRef.
- S. J. Sun, J. Xu, S. G. Dai and X. Han, Biomed. Environ. Sci., 2006, 19, 409 Search PubMed.
- H. Zhong and W. X. Wang, Environ. Sci. Technol., 2006, 40, 3794 CrossRef CAS.
- M. O. Mbila, M. L. Thompson, J. S. C. Mbagwu and D. A. Laird, J. Environ. Qual., 2001, 30, 1667 CAS.
- I. Ahumada, P. Escudero, L. Ascar, J. Mendoza and P. Richter, Commun. Soil Sci. Plant Anal., 2004, 35, 1615 CrossRef CAS.
- Z. Y. Hseu, Soil Sci., 2006, 171, 341 CrossRef CAS.
- C. Brunori, C. Cremisini, L. D'Annibale, P. Massanisso and V. Pinto, Anal. Bioanal. Chem., 2005, 381, 1347 CrossRef CAS.
- A. M. Moreno, L. Perez and J. G. Parra, Chem. Speciation Bioavailability, 2005, 17, 11 Search PubMed.
- B. P. Cid, M. D. Gonzalez and E. F. Gomez, Analyst, 2002, 127, 681 RSC.
- O. Abollino, M. Aceto, M. Malandrino, E. Mentasti, C. Sarzanini and F. Petrella, Chemosphere, 2002, 49, 545 CrossRef CAS.
- M. Ramirez, S. Massolo, R. Frache and J. A. Correa, Mar. Pollut. Bull., 2005, 50, 62 CrossRef CAS.
- S. R. Cook and A. Parker, Water, Air, Soil Pollut., 2006, 170, 95 CrossRef CAS.
- S. Audry, G. Blanc and J. Schafer, Sci. Total Environ., 2006, 363, 216 CrossRef CAS.
- H. B. Jung, S. T. Yun, B. Mayer, S. O. Kim, S. S. Park and P. K. Lee, Environ. Geol., 2005, 48, 437 CrossRef CAS.
- S. O. Ndzangou, M. Richer-LaFleche and D. Houle, Appl. Geochem., 2006, 21, 2135 CrossRef CAS.
- S. I. Korfali and B. E. Davies, Adv. Environ. Res., 2004, 8, 599 Search PubMed.
- S. I. Korfali and B. E. Davies, Environ. Geochem. Health, 2005, 27, 385 Search PubMed.
- E. Galan, J. L. Gomez-Ariza, I. Gonzalez, J. C. Fernandez-Caliani, E. Morales and I. Giraldez, Appl. Geochem., 2003, 18, 409 CrossRef CAS.
- C. Kraft, W. von Tumpling and D. W. Zachmann, Acta Hydrochim. Hydrobiol., 2006, 34, 257 CrossRef CAS.
- J. R. Brydie and D. A. Polya, Mineral. Mag., 2003, 67, 289 CrossRef CAS.
- M. H. D. Pestana and M. L. L. Formoso, J. Phys. IV, 2003, 107, 1049 Search PubMed.
- L. J. Tsai, K. C. Yu, S. F. Chen, P. Y. Kung, C. Y. Chang and C. H. Lin, Water Res., 2003, 37, 4623 CrossRef CAS.
- E. Van Hullebusch, P. Chatenet, V. Deluchat, P. M. Chazal, D. Froissard, P. N. L. Lens and M. Baudu, J. Phys. IV, 2003, 107, 1333 Search PubMed.
- I. Ko, Y. Y. Chang, C. H. Lee and K. W. Kim, J. Hazard. Mater., 2005, 127, 1 CrossRef CAS.
- S. Tandy, K. Bossart, R. Mueller, J. Ritschel, L. Hauser, R. Schulin and B. Nowack, Environ. Sci. Technol., 2004, 38, 937 CrossRef CAS.
- C. Chaiyaraksa and N. Sriwiriyanuphap, Chemosphere, 2004, 56, 1129 CrossRef CAS.
- E. E. C. de Melo, C. W. A. do Nascimento and A. C. Q. Santos, Rev. Bras. Cienc. Solo, 2006, 30, 1051 Search PubMed.
- Y. G. Liu, X. H. Wang, G. M. Zeng, X. Li, C. H. Zhou, T. Fan, Y. L. Li and X. Z. Yuan, Pedosphere, 2006, 16, 312 Search PubMed.
- J. G. Shi, Z. X. Yuan, S. F. Zhang, G. H. Huang, F. Dai, J. B. Li, A. Chakma, X. S. Qin and H. L. Liu, Trans. Nonferrous Met. Soc. China, 2004, 14, 66 Search PubMed.
- H. Massara, C. N. Mulligan and J. Hadjinicolaou, Soil. Sediment Contam., 2007, 16, 1 Search PubMed.
- M. Vanthuyne, A. Maes and P. Cauwenberg, Miner. Eng., 2003, 16, 1131 CrossRef CAS.
- G. M. Nystrom, L. M. Ottosen and A. Villumsen, J. Phys. IV, 2003, 107, 975 Search PubMed.
- L. M. Ottosen, K. Lepkova and M. Kubal, J. Hazard. Mater., 2006, 137, 113 CrossRef CAS.
- X. J. Chen, Z. M. Shen, Y. M. Lei, B. X. Ju and W. H. Wang, Aust. J. Soil Res., 2006, 44, 523 Search PubMed.
- W. Friesl, E. Lombi, O. Horak and W. W. Wenzel, J. Plant Nutr. Soil Sci., 2003, 166, 191 CrossRef CAS.
- S. Penilla, F. Bordas and J. C. Bollinger, J. Colloid Interface Sci., 2005, 292, 20 CrossRef CAS.
- F. Garrido, V. Illera and M. T. Garcia-Gonzalez, Appl. Geochem., 2005, 20, 397 CrossRef CAS.
- J. L. Daniels and G. P. Das, Environ. Eng. Sci., 2006, 23, 42 CAS.
- J. Kumpiene, S. Ore, G. Renella, M. Mench, A. Lagerkvist and C. Maurice, Environ. Pollut., 2006, 144, 62 CrossRef CAS.
- J. Kumpiene, A. Lagerkvist and C. Maurice, Environ. Pollut., 2007, 145, 365 CrossRef CAS.
- J. C. Zwonitzer, G. M. Pierzynski and G. M. Hettiarachchi, Water, Air, Soil Pollut., 2003, 143, 193 CrossRef CAS.
- R. X. Cao, L. Q. Ma, M. Chen, S. P. Singh and W. G. Harris, Environ. Pollut., 2003, 122, 19 CrossRef CAS.
- M. Chen, L. Q. Ma, S. P. Singh, R. X. Cao and R. Melamed, Adv. Environ. Res., 2003, 8, 93 Search PubMed.
- S. B. Chen, Y. G. Zhu and Y. B. Ma, J. Hazard. Mater., 2006, 134, 74 CrossRef CAS.
- S. B. Chen, Y. G. Zhu, Y. B. Ma and G. McKay, Environ. Pollut., 2006, 139, 433 CrossRef CAS.
- L. J. Cajuste, L. Cajuste, C. García-Osorio and J. Cruz-Diaz, Commun. Soil Sci. Plant Anal., 2006, 37, 2541 CrossRef CAS.
- L. S. Forsberg and S. Ledin, Sci. Total Environ., 2006, 358, 21 CrossRef CAS.
- R. Clemente and M. P. Bernal, Chemosphere, 2006, 64, 1264 CrossRef CAS.
- B. P. Spalding, Environ. Sci. Technol., 2001, 35, 4327 CrossRef CAS.
- A. Fuentes, M. Llorens, J. Saez, M. I. Aguilar, J. F. Ortuno and V. F. Meseguer, J. Hazard. Mater., 2004, 108, 161 CrossRef CAS.
- B. Kim and M. B. McBride, Environ. Pollut., 2006, 144, 475 CrossRef CAS.
- A. R. A. Usman and A. Ghallab, Chem. Ecol., 2006, 22, 267 Search PubMed.
- R. Clemente, A. Escolar and M. P. Bernal, Bioresour. Technol., 2006, 97, 1894 CrossRef CAS.
- D. C. Su and J. W. C. Wong, Environ. Int., 2004, 29, 895 CrossRef CAS.
- C. Oliveira, N. Sobrinho and N. Mazur, Rev. Bras. Cienc. Solo, 2003, 27, 171 Search PubMed.
- S. Li, R. L. Liu, M. Wang, X. B. Wang, H. Shan and H. T. Wang, Geoderma, 2006, 136, 260 CrossRef CAS.
- M. Rajaie, N. Karimian, M. Maftoun, J. Yasrebi and M. T. Assad, Geoderma, 2006, 136, 533 CrossRef CAS.
- M. Jalali and Z. V. Khanlari, Soil. Sediment Contam., 2006, 15, 603 Search PubMed.
- M. Antilen, N. Araya, M. Briceno and M. Escudey, Aust. J. Soil Res., 2006, 44, 619 Search PubMed.
- P. Castaldi, L. Santona and P. Melis, Fresenius Environ. Bull., 2006, 15, 1133 CAS.
- M. Turek, T. Korolewicz and J. Ciba, Soil. Sediment Contam., 2005, 14, 143 Search PubMed.
- A. T. Lombardi, O. J. Garcia and W. A. N. Menezes, World J. Microbiol. Biotechnol., 2006, 22, 1013 CrossRef CAS.
- C. Yuan and C. H. Weng, Chemosphere, 2006, 65, 88 CrossRef CAS.
- V. K. Tyagi, Geosynth. Int., 2006, 13, 47 Search PubMed.
- J. Y. Wang, D. S. Zhang, O. Stabnikova and J. H. Tay, J. Hazard. Mater., 2005, 124, 139 CrossRef CAS.
- T. Aarab, M. Smeyers, M. Remy, B. Godden and J. P. Delhaye, Waste Manage. (Amsterdam, Neth.), 2006, 26, 1024 Search PubMed.
- T. T. Lim, J. H. Tay, L. C. Tan, V. Choa and C. I. Teh, Environ. Geol., 2004, 47, 1 CrossRef CAS.
- T. T. Lim, J. Chu and M. H. Goi, Waste Manage. (Amsterdam, Neth.), 2006, 26, 1294 Search PubMed.
- Q. Y. Huang, W. L. Chen and X. J. Guo, Commun. Soil Sci. Plant Anal., 2004, 35, 947 CrossRef CAS.
- S. Eich-Greatorex and L. T. Strand, Soil Sci. Soc. Am. J., 2006, 70, 778 CrossRef CAS.
- M. Arienzo, P. Adamo and V. Cozzolino, Sci. Total Environ., 2004, 319, 13 CrossRef CAS.
- S. Ishikawa, N. Ae, M. Murakami and T. Wagatsuma, Soil Sci. Plant Nutr., 2006, 52, 32 Search PubMed.
- V. U. Ultra, A. Yano, K. Iwasaki, S. Tanaka, Y. M. Kang and K. Sakurai, Soil Sci. Plant Nutr., 2005, 51, 193 Search PubMed.
- R. Santamaria-Fernandez, M. R. Cave and S. J. Hill, J. Environ. Monit., 2003, 5, 929 RSC.
- Y. B. Ma and N. C. Uren, Nutr. Cycling Agroecosyst., 2006, 76, 11 Search PubMed.
- R. B. Herbert, J. Geochem. Explor., 2006, 90, 197 CrossRef CAS.
- B. Clozel, V. Ruban, C. Durand and P. Conil, Appl. Geochem., 2006, 21, 1781 CrossRef CAS.
- K. Rasa, T. Peltovuori and H. Hartikainen, Sci. Total Environ., 2006, 366, 819 CrossRef CAS.
- M. E. Soltan, Chem. Ecol., 2006, 22, 359 Search PubMed.
- C. S. C. Wong, S. C. Wu, N. S. Duzgoren-Aydin, A. Aydin and M. H. Wong, Environ. Pollut., 2007, 145, 434 CrossRef CAS.
- J. L. Luque-Garcia and L. de Castro, TrAC, Trends Anal. Chem., 2003, 22, 41 CrossRef CAS.
- B. Perez-Cid, I. Lavilla and C. Bendicho, Anal. Chim. Acta, 1998, 360, 35 CrossRef CAS.
- B. Perez-Cid, I. Lavilla and C. Bendicho, Fresenius’ J. Anal. Chem., 1999, 363, 667 CrossRef CAS.
- B. Perez-Cid, I. Lavilla and C. Bendicho, Int. J. Environ. Anal. Chem., 1999, 73, 79 CrossRef CAS.
- F. S. Sun, Z. Y. Zhan, K. S. Zhang and Y. Wang, J. Environ. Sci. (China), 2004, 16, 957 Search PubMed.
- A. Vaisanen and A. Kiljunen, Int. J. Environ. Anal. Chem., 2005, 85, 1037 CrossRef.
- C. M. Davidson and G. Delevoye, J. Environ. Monit., 2001, 3, 398 RSC.
- A. Marin, A. Lopez-Gonzalvez and C. Barbas, Anal. Chim. Acta, 2001, 442, 305 CrossRef CAS.
- I. Ipolyi, C. Brunori, C. Cremisini, P. Fodor, L. Macaluso and R. Morabito, J. Environ. Monit., 2002, 4, 541 RSC.
- G. M. Greenway and Q. J. Song, J. Environ. Monit., 2002, 4, 950 RSC.
- S. Canepari, E. Cardarelli, S. Ghighi and L. Scimonelli, Talanta, 2005, 66, 1122 CrossRef CAS.
- B. Krasnodebska-Ostrega, M. Kaczorowska and J. Golimowski, Microchim. Acta, 2006, 154, 39 CrossRef CAS.
- A. Vaisanen, R. Suontamo, J. Silvonen and J. Rintala, Anal. Bioanal. Chem., 2002, 373, 93 CrossRef.
- M. Gulmini, G. Ostacoli, V. Zelano and A. Torazzo, Analyst, 1994, 119, 2075 RSC.
- B. Perez-Cid, I. Lavilla and C. Bendicho, Anal. Chim. Acta, 1999, 378, 201 CrossRef CAS.
- B. Gumgum and G. Ozturk, Chem. Speciation Bioavailability, 2001, 13, 25 Search PubMed.
- M. Dutta, K. Chandrasekhar, S. Dutta and A. K. Das, At. Spectrosc., 2005, 26, 137 CAS.
- M. Dutta and A. K. Das, J. Indian Chem. Soc., 2005, 82, 433 CAS.
- P. Pazos-Capeans, M. C. Barciela-Alonso, A. Bermejo-Barrera and P. Bermejo-Barrera, Talanta, 2005, 65, 678 CrossRef CAS.
- M. Dutta, K. Chandrasekhar and A. K. Das, At. Spectrosc., 2005, 26, 14 CAS.
- E. Campos, E. Barahona, M. Lachica and M. D. Mingorance, Anal. Chim. Acta, 1998, 369, 235 CrossRef CAS.
- K. L. Laban and B. P. Atkin, Int. J. Coal Geol., 1999, 41, 351 CrossRef CAS.
- B. Perez Cid, A. Fernandez Albores, E. Fernandez Gomez and E. Falque Lopez, Anal. Chim. Acta, 2001, 431, 209 CrossRef CAS.
- B. Perez Cid, A. Fernandez Albores, E. Fernandez Gomez and E. Falque Lopez, Analyst, 2001, 126, 1304 RSC.
- R. Santamaria-Fernandez, M. R. Cave and S. J. Hill, Anal. Chim. Acta, 2006, 557, 344 CrossRef CAS.
- R. Santamaria-Fernandez, A. Moreda-Pineiro and S. J. Hill, J. Environ. Monit., 2002, 4, 330 RSC.
- M. R. Cave, A. E. Milodowski and E. N. Friel, Geochem.: Explor., Environ., Anal., 2004, 4, 71 CrossRef CAS.
- D. Beauchemin, K. Kyser and D. Chipley, Anal. Chem., 2002, 74, 3924 CrossRef CAS.
- M. Jimoh, W. Frenzel and V. Muller, Anal. Bioanal. Chem., 2005, 381, 438 CrossRef CAS.
- R. Chomchoei, M. Miro, E. H. Hansen and J. Shiowatana, Anal. Chem., 2005, 77, 2720 CrossRef CAS.
- R. Chomchoei, M. Miro, E. H. Hansen and J. Shiowatana, Anal. Chim. Acta, 2005, 536, 183 CrossRef CAS.
- P. S. Fedotov, E. Y. Savonina, R. Wennrich and B. Y. Spivakov, Analyst, 2006, 131, 509 RSC.
- P. S. Fedotov, R. Wennrich, H. J. Stark and B. Y. Spivakov, J. Environ. Monit., 2005, 7, 22 RSC.
- P. S. Fedotov, A. G. Zavarzina, B. Y. Spivakov, R. Wennrich, J. Mattusch, K. D. C. Titze and V. V. Demin, J. Environ. Monit., 2002, 4, 318 RSC.
- O. N. Katasonova, P. S. Fedotov, V. K. Karandashev and B. Y. Spivakov, J. Anal. Chem., 2005, 60, 684 CrossRef CAS.
- E. Y. Savonina, P. S. Fedotov and R. Wennrich, J. Anal. Chem., 2006, 61, 702 CrossRef CAS.
- J. Buanuam, K. Tiptanasup, J. Shiowatana, M. Miro and E. H. Hansen, J. Environ. Monit., 2006, 8, 1248 RSC.
- T. Nakazato, M. Akasaka and H. Tao, Anal. Bioanal. Chem., 2006, 386, 1515 CrossRef CAS.
- R. Chomchoei, J. Shiowatana and P. Pongsakul, Anal. Chim. Acta, 2002, 472, 147 CrossRef CAS.
- J. Buanuam, J. Shiowatana and P. Pongsakul, J. Environ. Monit., 2005, 7, 778 RSC.
- N. Tongtavee, J. Shiowatana and R. G. McLaren, Int. J. Environ. Anal. Chem., 2005, 85, 567 CrossRef CAS.
- N. Tongtavee, J. Shiowatana, R. G. McLaren and J. Buanuam, Commun. Soil Sci. Plant Anal., 2005, 36, 2839 CrossRef CAS.
- M. Miro, E. H. Hansen, R. Chomchoei and W. Frenzel, TrAC, Trends Anal. Chem., 2005, 24, 759 CrossRef CAS.
- A. Barona and F. Romero, Soil Technol., 1996, 8, 303 CrossRef.
- M. H. W. Lam, A. Y. W. Tjia, C. C. Chan, W. P. Chan and W. S. Lee, Mar. Pollut. Bull., 1997, 34, 949 CrossRef CAS.
- I. Stanimirova, K. Zehl, D. L. Massart, Y. V. Heyden and J. W. Einax, Anal. Bioanal. Chem., 2006, 385, 771 CrossRef CAS.
- C. Sarbu, K. Zehl and J. W. Einax, Chemom. Intell. Lab. Syst., 2007, 86, 121 CrossRef CAS.
- B. Daus and H. W. Zwanziger, Fresenius’ J. Anal. Chem., 1995, 352, 444 CAS.
- J. Virkanen, Mar. Pollut. Bull., 1998, 36, 729 CrossRef CAS.
- K. C. Yu, L. J. Tsai, S. H. Chen, D. J. Chang and S. T. Ho, J. Environ. Sci. Health, Part A, 2001, 36, 1 Search PubMed.
- H. Sanei, F. Goodarzi and E. Van der Flier-Keller, J. Environ. Monit., 2001, 3, 27 RSC.
- L. J. Tsai, K. C. Yu, J. S. Huang and S. T. Ho, J. Environ. Sci. Health, Part A, 2002, 37, 1421 Search PubMed.
- S. Tokalioglu and S. Kartal, Chem. Anal. (Warsaw), 2002, 47, 627 CAS.
- J. S. Chang, K. C. Yu, L. J. Tsai and S. T. Ho, Water Sci. Technol., 1998, 38, 159 CrossRef CAS.
- Z. Borovec, Sci. Total Environ., 1996, 177, 237 CrossRef CAS.
- L. J. Tsai, K. C. Yu, J. S. Chang and S. T. Ho, Water Sci. Technol., 1998, 37, 217 CrossRef CAS.
- Y. W. Wang, C. G. Yuan, X. L. Jin and G. B. Jiang, J. Environ. Sci. (China), 2005, 17, 540 Search PubMed.
- R. Zufiaurre, A. Olivar, P. Chamorro, C. Nerin and A. Callizo, Analyst, 1998, 123, 255 RSC.
- M. R. Cave and J. Wragg, Analyst, 1997, 122, 1211 RSC.
- S. Tokalioglu and S. Kartal, Int. J. Environ. Anal. Chem., 2003, 83, 935 CrossRef CAS.
- W. Wilcke, M. Krauss and J. Kobza, J. Plant Nutr. Soil Sci., 2005, 168, 676 CrossRef CAS.
- N. Teutsch, Y. Erel, L. Halicz and A. Banin, Geochim. Cosmochim. Acta, 2001, 65, 2853 CrossRef CAS.
- J. R. Bacon, I. J. Hewitt and P. Cooper, J. Environ. Monit., 2004, 6, 766 RSC.
- X. D. Li, Z. G. Shen, O. W. H. Wai and Y. S. Li, Mar. Pollut. Bull., 2001, 42, 215 CrossRef CAS.
- P. M. Outridge, Water, Air, Soil Pollut., 2000, 118, 179 CrossRef CAS.
- B. Kober, M. Wessels, A. Bollhofer and A. Mangini, Geochim. Cosmochim. Acta, 1999, 63, 1293 CrossRef CAS.
- S. C. Wong, X. D. Li, G. Zhang, S. H. Qi and Y. S. Min, Environ. Pollut., 2002, 119, 33 CrossRef CAS.
- J. R. Bacon, J. G. Farmer, S. M. Dunn, M. C. Graham and S. I. Vinogradoff, Environ. Pollut., 2006, 141, 469 CrossRef CAS.
- A. Probst, L. Hernandez and J. L. Probst, J. Phys. IV, 2003, 107, 1103 Search PubMed.
- M. Steinmann and P. Stille, Appl. Geochem., 1997, 12, 607 CrossRef CAS.
- S. Emmanuel and Y. Erel, Geochim. Cosmochim. Acta, 2002, 66, 2517 CrossRef CAS.
- F. Monna, N. Clauer, T. Toulkeridis and J. R. Lancelot, Appl. Geochem., 2000, 15, 1291 CrossRef CAS.
- Y. Yokoo, T. Nakano, M. Nishikawa and H. Quan, Chem. Geol., 2004, 204, 45 CrossRef CAS.
- Y. F. Xu and F. Marcantonio, Geochim. Cosmochim. Acta, 2004, 68, 2649 CrossRef CAS.
- S. Emmanuel, Y. Erel, A. Matthews and N. Teutsch, Chem. Geol., 2005, 222, 23 CrossRef CAS.
- A. Sahuquillo, G. Rauret, A. Rehnert and H. Muntau, Anal. Chim. Acta, 2003, 476, 15 CrossRef CAS.
- S. Van Herreweghe, R. Swennen, C. Vandecasteele and V. Cappuyns, Environ. Pollut., 2003, 122, 323 CrossRef.
- M. Mihaljevic, M. Ponavic, V. Ettler and O. Sebek, Anal. Bioanal. Chem., 2003, 377, 723 CrossRef CAS.
- K. A. Hudson-Edwards, S. L. Houghton and A. Osborn, TrAC, Trends Anal. Chem., 2004, 23, 745 CrossRef CAS.
- J. A. Jay, N. K. Blute, K. Lin, D. Senn, H. F. Hemond and J. L. Durant, Environ. Sci. Technol., 2005, 39, 9174 CrossRef CAS.
- R. Donahue and M. J. Hendry, Appl. Geochem., 2003, 18, 1733 CrossRef CAS.
- J. Y. Kim, A. P. Davis and K. W. Kim, Environ. Sci. Technol., 2003, 37, 189 CrossRef CAS.
- P. K. Pandey, S. Nair, A. Bhui and M. Pandey, Curr. Sci., 2004, 86, 190 CAS.
- P. Bhattacharya, M. Claesson, J. Bundschuh, O. Sracek, J. Fagerberg, G. Jacks, R. A. Martin, A. D. Storniolo and J. M. Thir, Sci. Total Environ., 2006, 358, 97 CrossRef CAS.
- Z. I. Gonzalez, M. Krachler, A. K. Cheburkin and W. Shotyk, Environ. Sci. Technol., 2006, 40, 6568 CrossRef.
- R. X. Guo, J. L. Yang and Z. Y. Liu, Fuel Process. Technol., 2004, 85, 903 CrossRef CAS.
- I. Ko, J. S. Ahn, Y. S. Park and K. W. Kim, Chem. Speciation Bioavailability, 2003, 15, 67 Search PubMed.
- C. Patinha, E. F. da Silva and E. C. Fonseca, J. Geochem. Explor., 2004, 84, 167 CAS.
- N. Lihareva, Microchem. J., 2005, 81, 177 CrossRef CAS.
- J. S. Ahn, Y. S. Park, J. Y. Kim and K. W. Kim, Environ. Geochem. Health, 2005, 27, 147 Search PubMed.
- J. Y. Kim, K. W. Kim, J. Ahn, I. Ko and C. H. Lee, Environ. Geochem. Health, 2005, 27, 193 Search PubMed.
- A. G. Gault, D. R. Cooke, A. T. Townsend, J. M. Charnock and D. A. Polya, Sci. Total Environ., 2005, 345, 219 CrossRef CAS.
- M. Jang, J. S. Hwang, S. I. Choi and J. K. Park, Chemosphere, 2005, 60, 344 CrossRef CAS.
- J. T. van Elteren, Z. Slejkovec, I. Arcon and H. J. Glass, Environ. Pollut., 2006, 139, 477 CrossRef CAS.
- L. Romero, H. Alonso, P. Campano, L. Fanfani, R. Cidu, C. Dadea, T. Keegan, I. Thornton and M. Farago, Appl. Geochem., 2003, 18, 1399 CrossRef CAS.
- M. Damris, G. A. O'Brien, W. E. Price and B. E. Chenhall, J. Environ. Monit., 2005, 7, 621 RSC.
- J. U. Lee, S. W. Lee, K. W. Kim and C. H. Yoon, Environ. Geochem. Health, 2005, 27, 159 Search PubMed.
- J. M. Yi, J. S. Lee and H. T. Chon, in On the Convergence of Bio-, Information-, Environmental-, Energy-, Space- and Nano-Technologies, Pts 1 and 2, Trans Tech Publications Inc., Zurich, Switzerland, 2005, vol. 277–279, p. 503 Search PubMed.
- J. M. Park, J. S. Lee, J. U. Lee, H. T. Chon and M. C. Jung, J. Geochem. Explor., 2006, 88, 134 CrossRef CAS.
- M. A. Taggart, M. Carlisle, D. J. Pain, R. Williams, D. Osborn, A. Joyson and A. A. Meharg, Sci. Total Environ., 2004, 323, 137 CrossRef CAS.
- S. Cornu, D. Montagne and P. Conil, C. R. Geosci., 2004, 336, 1007 Search PubMed.
- V. Matera, I. Le Hecho, A. Laboudigue, P. Thomas, S. Tellier and M. Astruc, Environ. Pollut., 2003, 126, 51 CrossRef CAS.
- Y. Takahashi, R. Minamikawa, K. H. Hattori, K. Kurishima, N. Kihou and K. Yuita, Environ. Sci. Technol., 2004, 38, 1038 CrossRef CAS.
- S. Saha, K. Ghosh, I. Das and S. K. Sanyal, J. Indian Chem. Soc., 2005, 82, 364 CAS.
- W. J. Liu, Y. G. Zhu, Y. Hu, P. N. Williams, A. G. Gualt, A. A. Meharg, J. M. Charnock and F. A. Smith, Environ. Sci. Technol., 2006, 40, 5730 CrossRef CAS.
- R. Datta, D. Sarkar, S. Sharma and K. Sand, Sci. Total Environ., 2006, 372, 39 CrossRef CAS.
- C. E. Renshaw, B. C. Bostick, X. H. Feng, C. K. Wong, E. S. Winston, R. Karimi, C. L. Folt and C. Y. Chen, J. Environ. Qual., 2006, 35, 61 CrossRef CAS.
- L. L. Embrick, K. M. Porter, A. Pendergrass and D. J. Butcher, Microchem. J., 2005, 81, 117 CrossRef CAS.
- E. Smith, J. Smith and R. Naidu, Sci. Total Environ., 2006, 363, 175 CrossRef CAS.
- D. Sarkar, R. Datta and S. Sharma, Chemosphere, 2005, 60, 188 CrossRef CAS.
- M. I. S. Gonzaga, J. A. G. Santos and L. Q. Ma, Environ. Pollut., 2006, 143, 254 CrossRef.
- I. Ko, C. H. Lee, K. P. Lee, S. W. Lee and K. W. Kim, Environ. Prog., 2006, 25, 39 CrossRef CAS.
- N. Caille, S. Swanwick, F. J. Zhao and S. P. McGrath, Environ. Pollut., 2004, 132, 113 CrossRef CAS.
- S. Jessen, F. Larsen, C. B. Koch and E. Arvin, Environ. Sci. Technol., 2005, 39, 8045 CrossRef CAS.
- T. Mrak, Z. Slejkovec and Z. Jeran, Talanta, 2006, 69, 251 CrossRef CAS.
- F. X. Han, W. L. Kingery, H. M. Selim, P. D. Gerard, M. S. Cox and J. L. Oldham, Sci. Total Environ., 2004, 320, 51 CrossRef CAS.
- A. Sahuquillo, G. Rauret, M. Bianchi, A. Rehnert and H. Muntau, Anal. Bioanal. Chem., 2003, 375, 578 CAS.
- N. S. Bloom, E. Preus, J. Katon and M. Hiltner, Anal. Chim. Acta, 2003, 479, 233 CrossRef CAS.
- G. E. M. Hall and P. Pelchat, Geochem.: Explor., Environ., Anal., 2005, 5, 115 CrossRef CAS.
- R. Fernandez-Martinez and M. I. Rucandio, Anal. Bioanal. Chem., 2003, 375, 1089 CAS.
- Y. Han, H. M. Kingston, H. M. Boylan, G. M. M. Rahman, S. Shah, R. C. Richter, D. D. Link and S. Bhandari, Anal. Bioanal. Chem., 2003, 375, 428 CAS.
- C. S. Kim, N. S. Bloom, J. J. Rytuba and G. E. Brown, Environ. Sci. Technol., 2003, 37, 5102 CrossRef CAS.
- C. Sladek and M. S. Gustin, Appl. Geochem., 2003, 18, 567 CrossRef CAS.
- R. Fernandez-Martinez, J. Loredo, A. Ordonez and M. I. Rucandio, Sci. Total Environ., 2005, 346, 200 CrossRef CAS.
- A. J. Slowey, J. J. Rytuba and G. E. Brown, Environ. Sci. Technol., 2005, 39, 1547 CrossRef CAS.
- D. Kocman, M. Horvat and J. Kotnik, J. Environ. Monit., 2004, 6, 696 RSC.
- C. M. Neculita, G. J. Zagury and L. Deschenes, J. Environ. Qual., 2005, 34, 255 CAS.
- D. Kocman, N. S. Bloom, H. Akagi, K. Telmer, L. Hylander, V. Fajon, V. Jereb, R. Jacimovic, B. Smodis, J. R. Ikingura and M. Horvat, J. Environ. Manage., 2006, 81, 146 Search PubMed.
- G. L. Liu, J. Cabrera, M. Allen and Y. Cai, Sci. Total Environ., 2006, 369, 384 CrossRef CAS.
- F. X. Han, Y. Su, D. L. Monts, C. A. Waggoner and M. J. Plodinec, Sci. Total Environ., 2006, 368, 753 CrossRef CAS.
- G. J. Zagury, C. M. Neculita, C. Bastien and L. Deschenes, Environ. Toxicol. Chem., 2006, 25, 1138 CrossRef CAS.
- A. Bernaus, X. Gaona, D. van Ree and M. Valiente, Anal. Chim. Acta, 2006, 565, 73 CrossRef CAS.
- H. B. Shi, L. N. Liang, G. B. Jiang and X. L. Jin, Environ. Int., 2005, 31, 357 CrossRef.
- C. Trombini, D. Fabbri, M. Lombardo, I. Vassura, E. Zavoli and M. Horvat, Cont. Shelf Res., 2003, 23, 1821 CrossRef.
- J. Beldowski and J. Pempkowiak, Chemosphere, 2003, 52, 645 CrossRef CAS.
- L. Boszke, A. Kowalski, W. Szczucinski, G. Rachlewicz, S. Lorenc and J. Siepak, Environ. Geol., 2006, 51, 527 CrossRef CAS.
- Y. D. Wang, C. Stauffer, C. Keller and M. Greger, Plant Soil, 2005, 275, 67 Search PubMed.
- M. T. Wright, D. R. Parker and C. Amrhein, Environ. Sci. Technol., 2003, 37, 4709 CrossRef CAS.
- T. R. Kulp and L. M. Pratt, Geochim. Cosmochim. Acta, 2004, 68, 3687 CrossRef CAS.
- T. T. Lim and K. H. Goh, Chemosphere, 2005, 58, 91 CrossRef CAS.
- C. A. P. de Leon, K. DeNicola, M. M. Bayon and J. A. Caruso, J. Environ. Monit., 2003, 5, 435 RSC.
- Y. Nakamaru, K. Tagami and S. Uchida, Chemosphere, 2005, 58, 1347 CrossRef CAS.
- M. C. Wang and H. M. Chen, Chemosphere, 2003, 52, 585 CrossRef CAS.
- I. Hagarova, M. Zemberyova and D. Bajcan, Chem. Pap., 2005, 59, 93 Search PubMed.
- P. T. Zawislanski, S. M. Benson, R. Terberg and S. E. Borglin, Environ. Sci. Technol., 2003, 37, 2415 CrossRef CAS.
- M. Bujdos, A. Mul'ova, J. Kubova and J. Medved, Environ. Geol., 2005, 47, 353 CrossRef CAS.
- C. Lussier, V. Veiga and S. Baldwin, Environ. Geol., 2003, 44, 905 CrossRef CAS.
- H. R. Vongunten and P. Benes, Radiochim. Acta, 1995, 69, 1 CAS.
- M. K. Schultz, W. Burnett, K. G. W. Inn and G. Smith, J. Radioanal. Nucl. Chem., 1998, 234, 251 CAS.
- M. K. Schultz, K. G. W. Inn, Z. C. Lin, W. C. Burnett, G. Smith, S. R. Biegalski and J. Filliben, Appl. Radiat. Isot., 1998, 49, 1289 CrossRef CAS.
- M. K. Schultz, W. C. Burnett and K. G. W. Inn, J. Environ. Radioact., 1998, 40, 155 CrossRef CAS.
- J. A. Lucey, L. L. Vintro, D. Boust, P. I. Mitchell, A. Gouzy and L. Bowden, J. Environ. Radioact., 2007, 93, 63 CrossRef CAS.
- A. J. Plater, M. Ivanovich and R. E. Dugdale, Appl. Geochem., 1992, 7, 101 CrossRef CAS.
- A. Takeda, H. Tsukada, Y. Takaku, S. Hisamatsu and M. Nanzyo, Sci. Total Environ., 2006, 367, 924 CrossRef CAS.
- C. Galindo, L. Mougin, S. Fakhi, A. Nourreddine, A. Larnghari and H. Hannache, J. Environ. Radioact., 2007, 92, 41 CrossRef CAS.
- K. Bunzl, R. Kretner, P. Schramel, M. Szeles and R. Winkler, Geoderma, 1995, 67, 45 CrossRef CAS.
- P. Blanco, F. V. Tome and J. C. Lozano, Appl. Radiat. Isot., 2004, 61, 345 CrossRef CAS.
- P. Blanco, F. V. Tome and J. C. Lozano, J. Environ. Radioact., 2005, 79, 315 CrossRef CAS.
- M. S. Al-Masri, K. Al-Karfan, H. Khalili and M. Hassan, J. Environ. Radioact., 2006, 91, 103 CrossRef CAS.
- S. E. Howe, C. M. Davidson and M. McCartney, J. Anal. At. Spectrom., 1999, 14, 163 RSC.
- J. L. Aguado, J. P. Bolivar and R. Garcia-Tenorio, J. Environ. Radioact., 2004, 74, 117 CrossRef CAS.
- A. J. G. Santos, B. P. Mazzilli, D. I. T. Favaro and P. S. C. Silva, J. Environ. Radioact., 2006, 87, 52 CrossRef CAS.
- M. Trautmannsheimer, P. Schramel, R. Winkler and K. Bunzl, Environ. Sci. Technol., 1998, 32, 238 CrossRef CAS.
- D. Desideri, L. Feduzi, A. A. Meli and C. Roselli, J. Radioanal. Nucl. Chem., 2006, 267, 551 CrossRef CAS.
- K. Bunzl, H. Flessa, W. Kracke and W. Schimmack, Environ. Sci. Technol., 1995, 29, 2513 CAS.
- M. H. Lee, Y. Y. Yoon, S. B. Clark and S. E. Glover, Radiochim. Acta, 2004, 92, 671 CrossRef CAS.
- M. Puhakainen, I. Riekkinen, T. Heikkinen, T. Jaakkola, E. Steinnes, K. Rissanen, M. Suomela and H. Thorring, J. Environ. Radioact., 2001, 52, 17 CrossRef CAS.
- P. McDonald, J. V. I. Batlle, A. Bousher, A. Whittall and N. Chambers, Sci. Total Environ., 2001, 267, 109 CrossRef CAS.
- A. Abdelouas, B. Grambow, M. Fattahi, Y. Andres and E. Leclerc-Cessac, Sci. Total Environ., 2005, 336, 255 CrossRef CAS.
- A. Baumann, W. Schimmack, H. Steindl and K. Bunzl, Radiat. Environ. Biophys., 1996, 35, 229 CrossRef CAS.
- M. M. Vinichuk, K. J. Johanson, K. Rosen and I. Nilsson, J. Environ. Radioact., 2005, 78, 77 CAS.
- M. J. Keith-Roach, K. Morris and H. Dahlgaard, Mar. Chem., 2003, 81, 149 CrossRef CAS.
- K. S. Davies and G. Shaw, Sci. Total Environ., 1993, 132, 71 CrossRef CAS.
- T. Ohnuki, J. Nucl. Sci. Technol., 1992, 29, 996 Search PubMed.
- A. Rigol, M. Roig, M. Vidal and G. Rauret, Environ. Sci. Technol., 1999, 33, 887 CrossRef CAS.
- K. Bunzl, W. Kracke, W. Schimmack and L. Zelles, J. Environ. Radioact., 1998, 39, 55 CrossRef CAS.
- T. T. Vandergraaf, D. J. Drew, D. Archambault and K. V. Ticknor, J. Contam. Hydrol., 1997, 26, 83 CrossRef CAS.
- B. Salbu and E. Steinnes, Analyst, 1992, 117, 243 RSC.
- D. H. Oughton, B. Salbu, G. Riise, H. Lien, G. Ostby and A. Noren, Analyst, 1992, 117, 481 RSC.
- B. Salbu, D. H. Oughton, A. V. Ratnikov, T. L. Zhigareva, S. V. Kruglov, K. V. Petrov, N. V. Grebenshakikova, S. K. Firsakova, N. P. Astasheva, N. A. Loshchilov, K. Hove and P. Strand, Health Phys., 1994, 67, 518 CAS.
- V. A. Poiarkov, A. N. Nazarov and N. N. Kaletnik, J. Environ. Radioact., 1995, 26, 259 CrossRef CAS.
- S. V. Krouglov, A. D. Kurinov and R. M. Alexakhin, J. Environ. Radioact., 1998, 38, 59 CrossRef CAS.
- Y. Sanada, T. Matsunaga, N. Yanase, S. Nagao, H. Amano, H. Takada and Y. Tkachenko, Appl. Radiat. Isot., 2002, 56, 751 CrossRef CAS.
- M. Vidal and G. Rauret, Int. J. Environ. Anal. Chem., 1993, 51, 85 CrossRef CAS.
- C. M. Davidson, M. D. Gibson, E. Hamilton, B. H. MacGillivray, J. Reglinski and E. Rezabal, Chemosphere, 2005, 58, 793 CrossRef CAS.
- B. Wilson and F. B. Pyatt, Sci. Total Environ., 2006, 370, 401 CrossRef CAS.
- P. Matus, J. Kubova, M. Bujdos and J. Medved, Talanta, 2006, 70, 996 CrossRef CAS.
- P. Matus, J. Kubova, M. Bujdos, V. Stresko and J. Medved, Anal. Bioanal. Chem., 2004, 379, 96 CAS.
- C. A. Lucho-Constantino, F. Prieto-Garcia, L. M. Del Razo, R. Rodriguez-Vazquez and H. M. Poggi-Varaldo, Agric. Ecosyst. Environ., 2005, 108, 57 CrossRef CAS.
- L. Wang, N. Kubota, T. Higashi and T. Fujimura, Soil Sci. Plant Nutr., 2004, 50, 339 Search PubMed.
- J. Poledniok and F. Buhl, Talanta, 2003, 59, 1 CrossRef CAS.
- Y. W. Chen, T. L. Deng, M. Filella and N. Belzile, Environ. Sci. Technol., 2003, 37, 1163 CrossRef CAS.
- E. Fuentes, H. Pinochet, M. Potin-Gautier and I. De Gregori, J. AOAC Int., 2004, 87, 60 CAS.
- F. Reith and D. C. McPhail, Geochim. Cosmochim. Acta, 2006, 70, 1421 CrossRef CAS.
- H. Al-Najar, A. Kaschl, R. Schulz and V. Romheld, Int. J. Phytorem., 2005, 7, 55 Search PubMed.
- D. Fliegel, Z. Berner, D. Eckhardt and D. Stuben, Anal. Bioanal. Chem., 2004, 379, 131 CrossRef CAS.
Footnote |
| † This review is dedicated to Allan Ure (see ref. 1) who was instrumental in the development of the BCR sequential extraction procedure and the preparation and certification of reference materials. Allan was involved in the work of various Royal Society of Chemistry analytical chemistry committees and European BCR programmes. |
| This journal is © The Royal Society of Chemistry 2008 |