2,5-Dimethylphenacyl carbamate: a photoremovable protecting group for amines and amino acids
Laxminarayana
Kammari
a,
Lukáš
Plíštil
a,
Jakob
Wirz
b and
Petr
Klán
*a
aDepartment of Organic Chemistry, Faculty of Science, Masaryk University, Kotlarska 2, CZ-611 37, Brno, Czech Republic. E-mail: klan@sci.muni.cz; Fax: +420-549492688; Tel: +420-549494856
bDepartement Chemie der Universität Basel, Klingelbergstrasse 80, CH-4056, Basel, Switzerland. E-mail: J.Wirz@unibas.ch; Fax: +41-61-2673855; Tel: +41-61-2673842
First published on 6th November 2006
Abstract
2,5-Dimethylphenacyl (DMP) carbamates (1a–c) released the corresponding free amines or amino acids in high chemical yields, albeit with quantum yields Φ of only 0.04–0.09, upon irradiation in either aprotic or protic solvents. The photoreaction proceeded principally from the triplet excited state via the E-photoenol. The lifetimes of the triplet enol and the E- and Z-enols in the ground state were determined by laser flash photolysis. The primary photoinitiated transformation liberated a carbamic acid derivative, which subsequently decarboxylated to the amino group-containing compound. Exhaustive irradiation of a DMP-protected aniline (1a) in acetonitrile did not provide aniline in quantitative chemical yields, because it was involved in reductive cleavage of the starting material as an electron donor, thereby decreasing the overall deprotection yield (86%). Phenylalanine methyl ester, liberated from 1c, was, however, obtained in excellent chemical yield (97%). It was also found that the carbamates, while thermally stable, released amines with higher quantum yields in acidic methanol solutions. The DMP chromophore is proposed as an excellent photoremovable protecting group for amino acids and, under specific conditions, for amines in organic synthesis and biochemistry.
Introduction
Photoremovable protecting groups (PPG) represent a useful tool for organic synthesis and biochemical applications.1–3 One current task is to develop strategies for the release of poor leaving groups such as alcohols or amines.2 Several studies have already described protection of amines as carbamoyl derivatives of photolabile 6-nitroveratryloxycarbonyl,4,5 benzoins,6–10o-nitrobenzyl,4,11,12 9-fluorenone,13 or brominated 7-hydroxycoumarin-4-ylmethyl14 groups. In general, an amino compound is released in two consecutive steps with initial photochemical formation of a carbamate anion followed by thermal decarboxylation to yield the corresponding free amine.15We have recently reported the photochemical behaviour and applications of the 2,5-dimethylphenacyl (DMP) protecting group for carboxylic acids,16,17 phosphates, sulfonates,18 and alcohols.19 The photochemistry of DMP derivatives is initiated by photoenolization and no further reagents are required to release a functionality.20–22 Here we report that the DMP photoremovable group can be utilized to protect amines and amino acids.
Results and discussion
Two amines (aniline and piperidine) and one amino acid derivative (phenylalanine methyl ester) were protected as DMP carbamates (1a–c). They were prepared by coupling the 2,5-dimethyl phenacyl chloroformate (3), synthesized from 2,5-dimethylphenacyl alcohol (2) and bis(trichloromethyl) carbonate (BTC),23 and the corresponding amino group-containing compound 1a–c in reasonable overall yields (42–68%) and high purity (Scheme 1). BTC,24 a stable crystalline solid utilized in this synthetic procedure, was significantly easier and safer to handle than the more commonly used phosgene.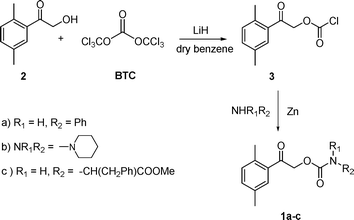 | ||
| Scheme 1 Synthesis of 1a–c. | ||
Irradiation of the carbamates 1a–c in methanol at 313 nm led to formation of the corresponding free amines, along with 2-(methoxymethyl)-5-methylacetophenone (4), 6-methyl-1-indanone (5), and carbon dioxide as the byproducts (Scheme 2). Indanone 5, in addition to CO2, was produced exclusively in cyclohexane or acetonitrile. This behaviour is analogous to that observed for DMP carboxylates or carbonates,17,19 except that the quantum yields for the carbamate disappearance were rather lower (Φ < 0.1) (Table 1). For comparison, while DMP carbonates afforded relatively low Φ in methanol (0.1–0.2), it reached 0.5 in non-polar cyclohexane. The product concentration ratio ([4]/[5]) was found to be 1.4 in methanol, which is similar to that obtained by photolysis of DMP carboxylates.17 Preparative (exhaustive) irradiation of 1c in acetonitrile, followed by purification, afforded phenylalanine methyl ester in 97% chemical yield. Photolysis of 1a and 1b in acetonitrile gave the corresponding amines in ∼85% yield (HPLC) and the reaction mixture contained an additional side product, 2,5-dimethylacetophenone.
| Compound | Φ (cyclohexane) | Φ (acetonitrile) | Φ (methanol) |
|---|---|---|---|
| a Φ were measured using valerophenone (Φ = 0.30)25 as an actinometer at 313 ± 5 nm (optical bench). The concentration of the starting material was 2.0–3.5 × 10−3 M. The irradiated samples were analyzed using HPLC; each sample was measured at least three times and the standard deviation of the mean is indicated. | |||
| 1a | 0.054 ± 0.002 | 0.035 ± 0.001 | 0.027 ± 0.002 |
| 1b | 0.087 ± 0.001 | 0.069 ± 0.001 | 0.056 ± 0.004 |
| 1c | 0.089 ± 0.001 | 0.070 ± 0.002 | 0.061 ± 0.001 |
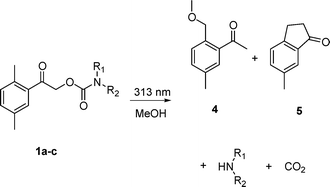 | ||
| Scheme 2 Photolysis of 1a–c in methanol. | ||
We have already demonstrated that photoenolization20,21 is the key reaction step in photochemical release of DMP protected functionalities.17–19,26 Investigation of 1a by laser flash photolysis indicated that photolysis in both cyclohexane and methanol produced three intermediates, one short-lived transient (λmax = 340 nm), the lifetime of which was sensitive to the presence of oxygen, and two longer-lived ones with λmax = 370 nm (Table 2). Based on previous studies,17,19 the first species was assigned to the triplet photoenol, 3E, and the second and third one to the Z- and E-xylylenols in the ground state (Scheme 3, Table 2). The former, shorter-lived transient possesses a geometry that allows for back hydrogen transfer–intramolecular reketonization.21 The quantum yield of the phototransformation of 1a was reduced upon addition of the triplet quencher piperylene in both cyclohexane and methanol (Fig. 1). While product formation was completely suppressed in methanol, approximately 30% of the reaction was not quenched in cyclohexane at high piperylene concentrations. This indicates that indanone 5 is partially formed via the singlet pathway in non-polar solvents.
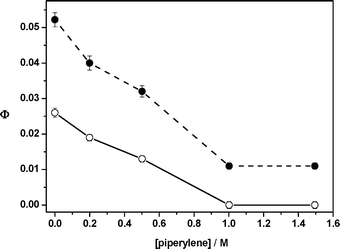 | ||
| Fig. 1 Photolysis of 1a in the presence of piperylene in methanol (○) and cyclohexane (●). | ||
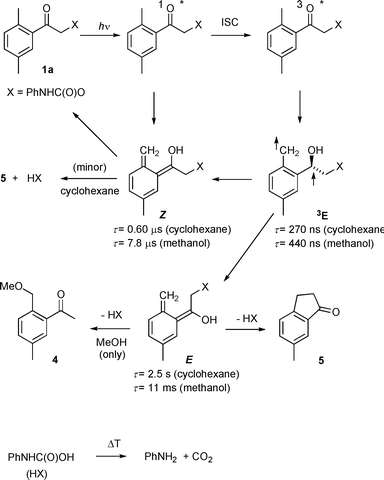 | ||
| Scheme 3 Photochemistry of 1a. | ||
The kinetic data listed in Table 2 are almost identical to those obtained for DMP carbonates,19 which are, along with carbamates, rather poor leaving groups. The release of such a moiety appears to be too slow to compete efficiently with intramolecular reketonization of the Z-photoenol (Scheme 3), which is the major photoproduct (about 90%). Indeed, the low quantum yields of the carbamate degradation (Table 1) demonstrate that ∼95% of the excitation energy is dissipated by processes restoring the starting material. In contrast, the elimination of better leaving groups, such as DMP chloride26 or DMP phosphoric and sulfonic esters,18 does compete with reketonization of the predominantly Z-photoenol and, hence, occurs primarily via this isomer despite its shorter lifetime. Subsequently, the carbamic acid, RNHC(O)OH, slowly decarboxylates in dark (e.g., τ1/2 ≈ 4.5 ms in aqueous solutions)9 to give free amino compounds. The decarboxylation rate varies with the pKa of the particular amino group but it seems to be comparable to that of the carbamate release. In comparison, N-1-(2-nitrophenyl)ethoxycarbonyl-caged glutamates and related carbamate-caged amino acids were found to be rate limited by decay of the aci-nitro intermediate, the process which generates the carbamate salt.27
As was mentioned before, exhaustive irradiation of 1a,b (in contrast to the protected phenylalanine 1c) in acetonitrile did not provide the corresponding amino compound in quantitative chemical yields. In addition to the major byproduct 5, formation of a small amount of 2,5-dimethylacetophenone (<10%) was observed. In order to clarify if free amines released from DMP carbamates can be responsible for any secondary photoreactions, a mixture of DMP benzoate and aniline was extensively irradiated in cyclohexane, affording 2,5-dimethylacetophenone in high chemical yields. This was expected because Falvey et al. have demonstrated that phenacyl esters reductively decompose into acetophenone and the corresponding carboxylic acids upon photolysis using good one-electron donors28–30 (Scheme 4). In a different study, Tanner et al. have found that the carboxylate anion is released from the anion radical with k = 108 s−1 in a one-electron reduction of phenacyl benzoate.31 It was, therefore, anticipated that such a reaction could efficiently compete with the photoenolization reaction of a DMP carbamate. When 1a was photolyzed in the presence of triethylamine (TEA) at 313 nm, where TEA does not absorb, the production of 2,5-dimethylacetophenone was the major chemical process observed (∼70%) at very high TEA concentrations (0.5 M). Two general mechanistic scenarios can therefore be anticipated:30 (1) sensitized reductive cleavage, where the DMP group accepts an electron from the excited (aromatic) amine, and (2) direct reductive cleavage, where the DMP group absorbs the light and then abstracts an electron from an amine. Furthermore, not only the starting material but also byproducts, such as indanone, can be involved in the subsequent electron transfer reactions, which lower the overall chemical deprotection yield. This fact diminishes the applicability of this group as PPG for simple amines, but the problem could be solved by choosing an appropriate irradiation wavelength or starting material concentration. Fortunately, no side reactions were observed in the case of 1c. Since phenylalanine has a higher adiabatic ionization energy (Ei = 8.5 eV32) than aliphatic amines (e.g., TEA, 7.5 eV33), the electron transfer reactions could not apparently compete with the photoenolization.
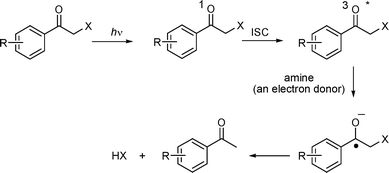 | ||
| Scheme 4 The photoinduced electron transfer deprotection of the phenacyl moiety.28 | ||
The ground state acid hydrolysis of carbamates has been shown by Lee et al. to proceed via a nucleophilic attack of water on the carbonyl carbon of an N-protonated precursor, although the carbonyl oxygen is also protonated at lower pH.34 As a result, an unstable zwitterionic form of the carbamic acid was shown to be a reaction intermediate.35 The protected amino acid 1c was quite stable toward dark acid-catalyzed hydrolysis in aqueous methanol ( 10% water) above pH 1.5 at 20 °C. When irradiated, the quantum yields of the phenylalanine methyl ester release increased in acidic (HCl) solutions. For example, Φ was higher by a factor of 3 at pH 1.5 than in a neutral solution (Fig. 2). The maximum quantum yields reached the values obtained in the photolysis of DMP carbonates19 or carboxylates17 in methanol. The leaving group ability of carbamic acid derivatives is expected to be comparable to or lower than those of carbonic acid (pKa = 6.4) or simple aliphatic carboxylic acids (pKa = 4.8). Both N- and O-protonation of a carbamate will unquestionably support the release of carbamic acid from xylylenols (Scheme 3) because the positive charge is located close to the bond being cleaved by heterolysis. Faster carbamate cleavage consequently decreases the probability that Z-photoenol would undergo back hydrogen transfer to the starting material. Indeed, at pH 2.5, the photochemical product formation from 1c was no longer completely quenched by piperylene addition but its yield was still reduced by 90% at high quencher concentrations (4 M). Therefore, the triplet state pathway, most probably involving the E-photoenol, still dominates in a polar acidic environment. Proton transfer quenching of the excited aromatic ketone36 is likely to compete with photoenolization at low pH. For example, the Norrish type II reaction of valerophenone is quenched at pH values below 2.37
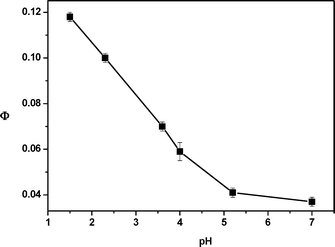 | ||
| Fig. 2 The quantum yields of 1c degradation in aqueous methanol (10% water) as a function of apparent pH (as measured with a glass electrode). | ||
Finally, the phenylalanine derivative 8, doubly DMP-protected on both the carboxylic and amino functional groups, was synthesized in order to compare simultaneous deprotection efficiencies from two different reaction sites of one molecule. Its synthesis started from N-Boc-DMP phenylalanate (6), obtained from DMP chloride and Boc-phenylalanine in the presence of DBU. The Boc group was then removed and the monoprotected compound 7 underwent a substitution reaction with 2,5-dimethylphenacyl chloroformate (3) in the presence of Zn powder to yield 8 (Scheme 5). Irradiation of 8 in acetonitrile at 313 nm (Scheme 6) afforded free phenylalanine with an apparent quantum yield of about 0.08, which corresponds well to that obtained for a monoprotected phenylalanine 1c degradation. The initial quantum yield of the carboxylic group specific release was higher (∼0.13), similar to that obtained for DMP carboxylates.17 However, the monoprotected compound 9 was never obtained as a sole product even at very low conversions.
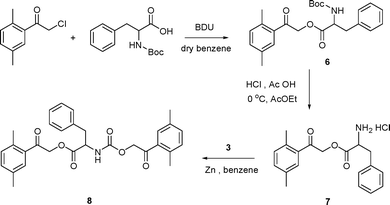 | ||
| Scheme 5 Synthesis of 8. | ||
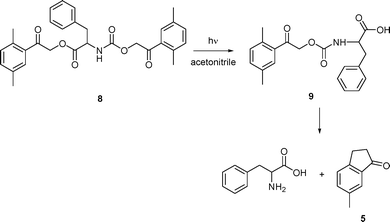 | ||
| Scheme 6 Photolysis of 8. | ||
Experimental
Materials and methods
The solvents were purified by distillation through a vacuum-sealed column (70 cm) packed with glass detritus. Bis(trichloromethyl) carbonate (BTC) (99%) and Boc-L-phenylalanine (>99%) were obtained from Aldrich, and 1,8-diazabicyclo[5.4.0]undec-7-ene (BDU) (98%) from Arcos Organics. NMR spectra were determined on a Bruker 300 MHz spectrometer. 1H and 13C NMR data were measured in CDCl3. The assignment of the 13C NMR spectra was based on an APT technique. Mass spectra were recorded on a Fisons Instrument Trio 1000 spectrometer in positive mode with EI. HPLC analyses were performed on a Shimadzu LC-10 AD equipped with a C18 column and UV-Vis detection. A mixture of acetonitrile (Merck; HPLC grade) and water was used as the mobile phase. A Labio CPI 03 equipped with a MAG 5 column (Biospher PSI 100; 7 µm) was used in preparative liquid chromatography separations. Melting points were measured on a Kofler hot stage VEB Wägetechnik Rapido 79/2106. UV spectra were obtained on a Shimadzu UV-1601 instrument with matched 1.0 cm quartz cells.Synthetic procedures
Preparative photolysis
Free amines or amino acids were obtained in >86% yield using a standard irradiation protocol.17 Solutions of the carbamates in dry acetonitrile were irradiated with a 125 W medium pressure Hg lamp filtered through a Pyrex glass sleeve (λ >280 nm). The irradiation was stopped when the conversion reached 95% (HPLC). In the case of 1c, the resulting precipitate was dissolved in water, washed with aq. HCl (10%), and the product was separated by a normal phase preparative HPLC using dichloromethane as the mobile phase. The products were analyzed by HPLC and NMR, using the authentic compounds as the analytical standards.Quantum yield measurements
The experiments were carried out on an optical bench consisting of a high-pressure 200 W Hg lamp, an Oriel ConerStone 130 1/8 m monochromator with grating 200–1600 nm set to 313 ± 5 nm and a 1 cm quartz cell containing the sample solution degassed by purging with argon. The light intensity was monitored by a Si photodiode detector (UV enhanced) with an Oriel OPM multifunctional optical power meter and controlled by TRACQ32 software. Valerophenone was used as an actinometer.25In the electron transfer studies, the cyclohexane solutions of 1a (c = 2.6 × 10−3 M) containing triethylamine (c = 4.2 × 10−3–0.5 M) were irradiated at 313 ± 5 nm on an optical bench. In the study of the pH-dependent quantum yields at 313 ± 5 nm, the sample acidity was adjusted by adding a methanolic solution of HCl (0.01 M) to an aqueous methanol (10% of water) solution of 1c. The pH in the range of 1.5–7.0 was measured by a pH meter. Control experiments were carried out in the dark in order to determine the stability of the carbamate toward hydrolysis.
Laser flash photolysis measurements
Experiments were carried out by exciting the sample solutions with an absorbance of <0.5 at 248 nm using a KrF pulsed excimer laser (pulse energy ∼150 mJ, pulse duration ∼30 ns). A pulsed xenon arc was used as the monitoring beam (cell path length 4.5 cm, orthogonal to the excitation pulse). The monitoring light beam leaving the sample cell could be diverted either to a diode array behind a gated image intensifier, which enabled the measurement of absorbance difference spectra at selected time delays after excitation and with time windows down to 5 ns, or to a monochromator/photomultiplier unit to monitor absorbance changes over time at a given wavelength. The signal from the photomultiplier was amplified, digitized and analyzed by nonlinear least-squares fitting of a first order rate law. All solutions were degassed in three freeze–pump–thaw cycles and sealed under vacuum before the measurements to minimize quenching of transients by oxygen. To avoid interference from secondary photolysis, the samples were freshly prepared from cyclohexane or methanol stock solutions and discarded after each laser shot.Acknowledgements
We gratefully acknowledge financial support from the Czech Ministry of Education, Youth and Sport (MSM0021622413), the Grant Agency of the Czech Republic (203/05/0641), and the Swiss National Science Foundation (200020-105219/1). The authors express their thanks to Jaromir Literak for fruitful discussions.References
- R. S. Givens, P. G. Conrad, A. L. Yousef and J.-I. Lee, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. M. Horspool and F. Lenci, CRC, Boca Raton, FL, 2004, ch. 69pp. 1–46 Search PubMed.
- A. P. Pelliccioli and J. Wirz, Photoremovable protecting groups: reaction mechanisms and applications, Photochem. Photobiol. Sci., 2002, 1, 441–458 RSC.
- C. G. Bochet, Photolabile protecting groups and linkers, J. Chem. Soc., Perkin Trans. 1, 2002, 125–142 RSC.
- A. Patchornik, B. Amit and R. B. Woodward, Photosensitive protecting groups, J. Am. Chem. Soc., 1970, 92, 6333–6335 CrossRef CAS.
- C. G. Bochet, Wavelength-selective cleavage of photolabile protecting groups, Tetrahedron Lett., 2000, 41, 6341–6346 CrossRef CAS.
- J. F. Cameron, C. G. Willson and J. M. J. Frechet, New photolabile amino protecting groups-photogeneration of amines from (3′,5′-dimethoxybenzoinyl)oxy carbonyl carbamates, J. Chem. Soc., Chem. Commun., 1995, 923–924 RSC.
- J. F. Cameron, C. G. Willson and J. M. J. Frechet, Photogeneration of amines from α-keto carbamates: Photochemical studies, J. Am. Chem. Soc., 1996, 118, 12925–12937 CrossRef CAS.
- J. F. Cameron, C. G. Wilson and J. M. J. Frechet, Photogeneration of amines from α-keto carbamates: Design and preparation of photoactive compounds, J. Chem. Soc., Perkin Trans. 1, 1997, 2429–2442 RSC.
- G. Papageorgiou and J. E. T. Corrie, Synthesis and properties of carbamoyl derivatives of photolabile benzoins, Tetrahedron, 1997, 53, 3917–3932 CrossRef CAS.
- M. C. Pirrung and C. Y. Huang, Photochemical deprotection of 3′,5′-dimethoxybenzoin (DMB) carbamates derived from secondary amines, Tetrahedron Lett., 1995, 36, 5883–5884 CrossRef CAS.
- J. F. Cameron and J. M. J. Frechet, Photogeneration of organic bases from o-nitrobenzyl-derived carbamates, J. Am. Chem. Soc., 1991, 113, 4303–4313 CrossRef CAS.
- J. E. Beecher, J. F. Cameron and J. M. J. Frechet, Photogeneration of polymeric amines. Synthesis, photo-cross-linking and photoimaging of copolymers containing photoactive carbamate pendant groups, J. Mater. Chem., 1992, 2, 811–816 RSC.
- G. Bucher, J. C. Scaiano, R. Sinta, G. Barclay and J. Cameron, Laser flash photolysis of carbamates derived from 9-fluorenone oxime, J. Am. Chem. Soc., 1995, 117, 3848–3855 CrossRef CAS.
- T. Furuta, S. S. H. Wang, J. L. Dantzker, T. M. Dore, W. J. Bybee, E. M. Callaway, W. Denk and R. Y. Tsien, Brominated 7-hydroxycoumarin-4-ylmethyls: Photolabile protecting groups with biologically useful cross-sections for two photon photolysis, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1193–1200 CrossRef CAS.
- G. Papageorgiou, A. Barth and J. E. T. Corrie, Flash photolytic release of alcohols from photolabile carbamates or carbonates is rate-limited by decarboxylation of the photoproduct, Photochem. Photobiol. Sci., 2005, 4, 216–220 RSC.
- P. Klan, M. Zabadal and D. Heger, 2,5-Dimethylphenacyl as a new photoreleasable protecting group for carboxylic acids, Org. Lett., 2000, 2, 1569–1571 CrossRef CAS.
- M. Zabadal, A. P. Pelliccioli, P. Klan and J. Wirz, 2,5-Dimethylphenacyl esters: A photoremovable protecting group for carboxylic acids, J. Phys. Chem. A, 2001, 105, 10329–10333 CrossRef CAS.
- P. Klan, A. P. Pelliccioli, T. Pospisil and J. Wirz, 2,5-Dimethylphenacyl esters: A photoremovable protecting group for phosphates and sulfonic acids, Photochem. Photobiol. Sci., 2002, 1, 920–923 RSC.
- J. Literak, J. Wirz and P. Klan, 2,5-Dimethylphenacyl carbonates: A photoremovable protecting group for alcohols and phenols, Photochem. Photobiol. Sci., 2005, 4, 43–46 RSC.
- P. G. Sammes, Photoenolization, Tetrahedron, 1976, 32, 405–422 CrossRef CAS.
- R. Haag, J. Wirz and P. J. Wagner, The photoenolization of 2-methylacetophenone and related compounds, Helv. Chim. Acta, 1977, 60, 2595–2607 CrossRef CAS.
- P. J. Wagner and C. P. Chen, A rotation-controlled excited-state reaction. The photoenolization of ortho alkyl phenyl ketones, J. Am. Chem. Soc., 1976, 98, 239–241 CrossRef CAS.
- H. B. Shi, W. X. Hu and Y. Q. Sun, Preparation of chloroformates using bis(trichloromethyl) carbonate, J. Chem. Res. Synop., 2004, 708–709 Search PubMed.
- J. S. Yadav, G. S. Reddy, M. M. Reddy and H. M. Meshram, Zinc promoted simple and convenient synthesis of carbamates: An easy access for amino group protection, Tetrahedron Lett., 1998, 39, 3259–3262 CrossRef CAS.
- P. J. Wagner, I. E. Kochevar and A. E. Kemppainen, Type II photoprocesses of phenyl ketones. Procedures for determining meaningful quantum yields and triplet lifetimes, J. Am. Chem. Soc., 1972, 94, 7489–7494 CrossRef CAS.
- A. P. Pelliccioli, P. P. Klan, M. Zabadal and J. Wirz, Photorelease of HCl from o-methylphenacyl chloride proceeds through the Z-xylylenol, J. Am. Chem. Soc., 2001, 123, 7931–7932 CrossRef CAS.
- J. E. T. Corrie, A. Desantis, Y. Katayama, K. Khodakhah, J. B. Messenger, D. C. Ogden and D. R. Trentham, Postsynaptic activation at the squid giant synapse by photolytic release of L-glutamate from a caged L-glutamate, J. Physiol. (London), 1993, 465, 1–8 Search PubMed.
- A. Banerjee and D. E. Falvey, Protecting groups that can be removed through photochemical electron transfer: Mechanistic and product studies on photosensitized release of carboxylates from phenacyl esters, J. Org. Chem., 1997, 62, 6245–6251 CrossRef CAS.
- A. Banerjee, K. Lee, Q. Yu, A. G. Fang and D. E. Falvey, Protecting group release through photoinduced electron transfer: wavelength control through sensitized irradiation, Tetrahedron Lett., 1998, 39, 4635–4638 CrossRef CAS.
- D. E. Falvey and C. Sundararajan, Photoremovable protecting groups based on electron transfer chemistry, Photochem. Photobiol. Sci., 2004, 3, 831–838 RSC.
- D. D. Tanner, J. J. Chen, L. Chen and C. Luelo, Fragmentation of substituted acetophenone and halobenzophenone ketyls-calibration of a mechanistic probe, J. Am. Chem. Soc., 1991, 113, 8074–8081 CrossRef CAS.
- S. Campbell, E. M. Marzluff, M. T. Rodgers, J. L. Beauchamp, M. E. Rempe, K. F. Schwinck and D. L. Lichtenberger, Proton affinities and photoelectron-spectra of phenylalanine and N-methylphenylalanine and N,N-dimethylphenylalanine-correlation of lone-pair ionization energies with proton affinities and implications for N-methylation as a method to effect site-specific protonation of peptides, J. Am. Chem. Soc., 1994, 116, 5257–5264 CrossRef CAS.
- D. H. Aue, H. M. Webb and M. T. Bowers, Quantitative proton affinities, ionization potentials, and hydrogen affinities of alkylamines, J. Am. Chem. Soc., 1976, 98, 311–317 CrossRef CAS.
- I. Lee, C. K. Kim and B. C. Lee, Theoretical studies on the acid-hydrolysis of methyl carbamate, J. Comput. Chem., 1987, 8, 794–800 CrossRef CAS.
- S. L. Johnson and D. L. Morrison, Kinetics and mechanism of decarboxylation of N-arylcarbamates. Evidence for kinetically important zwitterionic carbamic acid species of short lifetime, J. Am. Chem. Soc., 1972, 94, 1323–1334 CrossRef CAS.
- M. Ramseier, P. Senn and J. Wirz, Photohydration of benzophenone in aqueous acid, J. Phys. Chem. A, 2003, 107, 3305–3315 CrossRef CAS.
- R. G. Zepp, M. M. Gumz, W. L. Miller and H. Gao, Photoreaction of valerophenone in aqueous solution, J. Phys. Chem. A, 1998, 102, 5716–5723 CrossRef CAS.
- A. Banerjee, K. Lee and D. E. Falvey, Photoreleasable protecting groups based on electron transfer chemistry. Donor sensitized release of phenacyl groups from alcohols, phosphates and diacids, Tetrahedron, 1999, 55, 12699–12710 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2007 |