Conformational analysis of the blue-light sensing protein YtvA reveals a competitive interface for LOV–LOV dimerization and interdomain interactions†
Valentina
Buttani
a,
Aba
Losi
*ab,
Thorsten
Eggert
c,
Ulrich
Krauss
c,
Karl-Erich
Jaeger
c,
Zhen
Cao
d and
Wolfgang
Gärtner
d
aDept. of Physics, University of Parma, via G.P. Usberti 7/A, 43100-Parma, Italy. E-mail: losia@fis.unipr.it
bCNR-INFM, Parma, Italy
cInstitut für Molekulare Enzymtechnologie, Heinrich-Heine Universität Düsseldorf, Forschungszentrum Jülich, D-52426 Jülich, Germany
dMax-Planck-Institut für Bioanorganische Chemie, Stiftstr. 34-36, 45470 Mülheim, Germany
First published on 27th October 2006
Abstract
The Bacillus subtilis protein YtvA is related to plant phototropins in that it senses UVA–blue-light by means of the flavin binding LOV domain, linked to a nucleotide-binding STAS domain. The structural basis for interdomain interactions and functional regulation are not known. Here we report the conformational analysis of three YtvA constructs, by means of size exclusion chromatography , circular dichroism (CD ) and molecular docking simulations . The isolated YtvA-LOV domain (YLOV, aa 25–126) has a strong tendency to dimerize, prevented in full-length YtvA, but still observed in YLOV carrying the N-terminal extension (N-YLOV, aa 1–126). The analysis of CD data shows that both the N-terminal cap and the linker region (aa 127–147) between the LOV and the STAS domain are helical and that the central β-scaffold is distorted in the LOV domains dimers. The involvement of the central β-scaffold in dimerization is supported by docking simulation of the YLOV dimer and the importance of this region is highlighted by light-induced conformational changes, emerging from the CD data analysis. In YtvA, the β-strand fraction is notably less distorted and distinct light-driven changes in the loops/turn fraction are detected. The data uncover a common surface for LOV–LOV and intraprotein interaction, involving the central β-scaffold, and offer hints to investigate the molecular basis of light-activation and regulation in LOV proteins.
Introduction
B. subtilis YtvA is a blue-light responsive protein (261 aa), carrying a flavin-binding LOV (light, oxygen, voltage) domain, with LOV belonging to the PAS (PerArntSim) superfamily.1 The photochemical LOV paradigm has emerged during the last years, thanks to the discovery and molecular characterization of phototropins (phot), blue-light receptors for a variety of responses in plants.2–4 Phot are organized in two N-terminal LOV domains (LOV1 and LOV2, ca. 110 amino acids) and a C-terminally located ser/thr kinase domain. A self-phosphorylation reaction5,6 is activated by UVA–blue light illumination of phot, thanks to the activation the LOV domains that carry a flavin-mononucleotide (FMN) as chromophore.7 Light activation of LOV domains triggers a photocycle that involves the reversible formation of a covalent adduct between a conserved cysteine residue and position C(4a) of FMN,7–11 formed upon decay of the FMN triplet state .12–14 Despite the similarities in the light-triggered reactions, the two phot-LOV domains have not the same functional significance, with the self-phosphorylation reaction being mostly mediated by LOV2,15,16 that also presents a larger quantum yield for the formation of the covalent adduct.9 The LOV paradigm is configuring as one of the most conserved among distant phyla, and LOV proteins are now well documented in eukaryotes and prokaryotes.17,18 In bacteria they are present in about 15–17% of the sequenced genomes, and light-induced, phot-like reactions have been demonstrated for some of them.18–20Although the mechanistic details of the photochemical FMN–Cys adduct formation are still under debate (see ref. 19 and references therein, and ref. 21) the photocycle of LOV domains is by far the best characterized part of the light-to-signal transduction chain, thanks to the fact that isolated LOV domains are readily expressed and have been functionally and structurally analyzed.9,10,22,23 A structure of full-length phot is not yet available and little is known about the mechanism leading to kinase activation and on the protein surfaces involved in domain–domain interactions. Light-driven unfolding of the helical linker connecting LOV2 and the kinase domain (Jα-linker) has been recently proposed to trigger the self-phosphorylation reaction, based on NMR spectroscopy and mutagenesis experiments.24,25 This idea has recently been challenged by the observation that the Jα-linker is not needed either for LOV2-kinase interaction or for light-driven phosphorylation of a heterologous substrate.15
In prokaryotes the LOV light-sensing module is coupled to diverse effector domains, such as kinases (similar to phot), phosphodiesterases, response regulators, DNA-binding transcription factors, regulators of stress sigma factors.17,18,26 Therefore, besides the intrinsic interest regarding their structure, function and physiological role, they also represent a powerful tool to understand fundamental and still open questions in the field of LOV-based photoperception. (i) Are the light-induced reactions, centered on the LOV domain, transmitted to effector partners by means of the same molecular mechanisms, and do LOV domains interact with partner domains by means of the same protein surface? In phot1-LOV2 the central β-scaffold has been demonstrated to participate in interdomain communication, making contact with the Jα-linker.24,25 A similar process has been observed with phot2-LOV2.27 (ii) Why only one LOV domain is present in bacterial LOV proteins, whereas phot possess two of such units organized in tandem?17,18 The LOV2 domain has a higher photocycle quantum yield than LOV1,9 and acts as the principal light-sensing domain triggering phot1 and phot2 kinase activity, whereas LOV1 might have a regulative role.16 The amino acid sequence of bacterial LOV domains has in general intermediate characteristics between LOV1 and LOV2.18 (iii) Which factors govern LOV–LOV dimerization, considered a key feature in PAS-mediated sensing/regulation,28,29 and which is the relevance of it during light sensing? It was shown by gel filtration chromatography that phot1-LOV1 has a tendency to dimerize, whereas LOV2 is monomeric.30 This has led to the suggestion that LOV1 is responsible for phot dimerization, providing a possible functional role for the tandem organization of LOV domains in phot.30 By means of pulsed thermal grating Terazima and coworkers, detected a transient volume increase (about 1.8 times, with time constant of 300 µs) during light activation of an extended phot1-LOV2 construct (including an N-terminal cap and the Jα-linker), and interpreted this phenomenon as a transient dimerization,31 whose functional significance is not known. Dimeric states have been detected by means of small-angle X-ray scattering (SAXS) for the LOV domain of FKF132 and phot LOV1 domains.33 The SAXS experiments showed that phot1-LOV2 is a dimer (in contrast with ref. 30 and 31) whereas phot2-LOV2 is monomeric.33 The LOV domain of WC-1 from Neurospora has also been shown to homodimerize in vitro.34
In this work we have investigated three different constructs of YtvA, in order to partially address these problems. In YtvA the LOV domain is linked to a C-terminal STAS domain (sulfate transportersantisigma-factor antagonists).35 This architecture is conserved in LOV proteins form other Firmicutes, e.g. in Listeria and Oceanobacillus genera.18 Recent work has shown that YtvA is a positive regulator in the environmental signaling pathway that activates the general stress factor σB36,37 and, most importantly, that the cysteine involved in the photoadduct formation is needed for its in vivo function,37 in turn regulated by blue-light activation.38 These last two recent studies allow to regard Ytva as a real flavin-based blue-light photoreceptor in B. subtilis, not only a blue-light sensitive protein. The STAS is thought to be the effector domain of YtvA, although little is known of its molecular functionality, with the exception that it confers to YtvA the ability to bind GTP and ATP,39 in analogy with another STAS protein.40 The constructs that we used are the LOV core (YLOV, aa 25–126), the N-YLOV comprising also the first 24 aa (aa 1–126) and the full-length protein YtvA. We applied gel filtration chromatography to detect possible dimers and circular dichroism spectroscopy for secondary structure determination, improving the data analysis with respect to previous work.41 The data uncover a common surface for YLOV homodimerization and interdomain interactions, and corroborate a molecular model of the YLOV dimer obtained by docking simulations . Similarities and differences with phot-LOV domains are discussed.
Experimental
Protein samples and chemicals
For the N-YLOV protein, the DNA sequence encoding LOV core + N-terminal cap (aa 1–126 of the full protein) was amplified by PCR (Polymerase Chain Reaction ). The recombinant plasmid of full-length YtvA in (pET28a) was used as template, the primers were:5′-CAGCCATATGGCTAGTTTTCAATCATT (forward)
5′-TATTACTCGAGTTAGGTGATATCATTCTGAATTC (reverse)
Platinum® Taq DNA Polymerase (Invitrogen, Karlsruhe, Germany) was used for the PCR. PCR product, digested with NdeI/XhoI (NEB, Ipswich, UK), was ligated into the expression vector pET28a (Novagen-Merck, Darmstadt, Germany), which was digested with the same restriction enzymes. An N-terminal extension, including the 6×His-tag (sequence: MGSSHHHHHHSSGLVPRGSH) was furnished, in the same way as for YtvA and YLOV. For the details of full-length His-tagged YtvA and its isolated LOV core (YLOV) generation, see previous reports.19,42 The His-tagged proteins were expressed in E. coli BL21 DE3 (Stratagene, Amsterdam, The Netherlands) using IPTG (BioMol, Hamburg, Germany) induction. The proteins were then purified by affinity chromatography on Talon (Qiagen, Hilden, Germany) and finally concentrated in Na-phosphate buffer 10 mM, NaCl 10 mM, pH = 8.
Chromatography
Gel filtration chromatography experiments were performed on a Pharmacia FPLC apparatus, using a Superdex 75 HR 10/30 column (Amersham Biosciences), equilibrated with Na-phosphate 10 mM, pH = 8, NaCl = 0.15 M. A calibration curve was made using bovine serum albumin (69 kDa), ovalbumin (42.7 kDa), α-chymotrypsin (25 kDa), myoglobin (16.9 kDa) and ribonuclease (13.7 kDa) (low Mw calibration kit, Amersham Biosciences). YtvA, YLOV and N-YLOV were loaded on the column at a concentration between 1 and 50 µM, to give a final concentration ranging from 0.05 to 2.5 µM at the detection peak, due to dilution through the column.Circular dichroism spectroscopy and data analysis
Circular dichroism (CD ) experiments were carried out using a Jasco J715 spectropolarimeter, calibrated with ammonium d-10-camporsulfonic acid. The measurements were carried out in the far-UV spectral region (195–240 nm) at a temperature of 20 °C and the buffer background was always subtracted. The optical pathlength was 0.2 cm. Protein concentration was estimated from the absorption coefficient at 220 nm, εYTVA220 = 492800 M–1 cm–1, εYLOV220 = 223900 M–1 cm–1 and εN-YLOV220 = 267900 M–1 cm–1, calculated by comparison with εFMN447 = 12500 M–1 cm–1, in a 1 : 1 protein to chromophore ratio (vide infra). The corresponding value for FMN is εFMN220 = 34500 M–1 cm–1, thus introducing a negligible error also in case that some apoprotein is present. The mean residue ellipticity ΘMRW was calculated from the concentration of residues, c, (281 aa for YtvA, 122 aa for YLOV and 146 aa for N-YLOV, including the tag, according to the formula ΘMRW = Θobs/(10 × cl), where c is in mol liter–1, l = 0.2 cm and Θobs is in mdeg and ΘMRW in deg cm2 dmol–1. Typically, the protein concentration was in the µM range and c = 10–6 × 281 = 2.8 × 10–4 mol liter–1 for YtvA, c = 10–6 × 122 = 1.2 × 10–4 mol liter–1 for YLOV and c = 10–6 × 146 = 1.4 × 10–4 mol liter–1 for N-YLOV. Prediction of secondary structure composition was performed using the convex constraint analysis (CCA) algorithm,43,44 and an extended curves dataset comprising 46 protein spectra.45 In CCA, the sum of the fractional weights of each component spectrum is constrained to be 1. In addition, a constraint-called volume minimization is defined which allows a finite number of component curves to be extracted from a set of spectra without relying on spectral nodes. CCA does not use X-ray crystallographic data in the deconvolution procedure. Once the basis curves are obtained, they must be assigned to specific secondary structures. The secondary structure was also predicted from the amino acid sequence by means of bioinformatic tools, using the consensus secondary structure prediction method at the Pôle Bioinformatique Lyonnais.46Docking simulation , evaluation of complexes and model validation
Docking simulations of the YLOV dimer were carried out at the ClusPro Server,47 using the DOT 1.048 and ZDOCK v. 2.349 programmes, employing the previously published YLOV structural model (PDB databank accession code 1IUM).19 The ClusPro docking algorithm evaluates billions of putative complexes, retaining a preset number with favourable surface complementarities. A filtering method is then applied to this set of structures, selecting those with good electrostatic and desolvation free energies for further clustering. Evaluation of the ClusPro predicted complexes was carried out using the VADAR tool,50 which calculates multi-chain parameters such as the accessible surface area and the percentage of accessible hydrophobic side chains. The quality of the ClusPro predicted YLOV dimer model was verified by using the protein structure analysis and validation server (SAVS) of the NIH MBI Laboratory for Structural Genomics and Proteomics at the University of California, Los Angeles (UCLA) (http://nihserver.mbi.ucla.edu/SAVS/) that implements the programs PROCHECK,51 WHAT_CHECK,52 ERRAT,53 VERIFY_3D,54 and PROVE.55 Additionaly, PPI-Pred56 and the computational interface alanine scanning tool at the Robetta server57 were used to predict the dimerization interface of the YLOV-monomer and of the YLOV dimers respectively.Results and discussion
Gel filtration chromatography
The elution profile of YLOV (Fig. 1) reveals that the majority of the protein is present in a state having Mw = 36.44 kDa, namely 2.63 times larger than the theoretical Mw (13.81 kDa, considering also the 20 aa at the N-terminal His-tag). This suggests a dimeric state that deviates from a spherical shape, as previously reported for other LOV domains.30,32,33 Traces of a globular monomer are observable in some preparations, as well as a small fraction of a larger aggregate, most probably a trimer, with Mw = 51.97 kDa. Light activation does not appreciably affect the elution profile of YLOV. N-YLOV is also mostly present in a dimeric state, with apparent Mw = 34.00 kDa, 2.06 times larger than the theoretical Mw (16.54 kDa, again including the His-tag). For N-YLOV the shape of the dimer is therefore approximately spherical. | ||
| Fig. 1 Elution profiles of the YLOV (dashed line) and N-YLOV (full line) domains of YtvA. The main peaks correspond to Mw = 2.6 × MwYLOV and Mw = 2.1 × MwN-YLOV, respectively. | ||
The full-length protein YtvA presents a larger heterogeneity and the elution profile is different among different preparations (Fig. 2). Up to three peaks can be identified, with peak 1 resulting in Mw = 72.85 kDa, peak 2 with Mw = 48.39 kDa and peak 3 with Mw = 35.07 kDa. The theoretical Mw of YtvA is 31.36 kDa, therefore we can assign the three peaks to a dimeric state (peak 1), an elongated monomer (peak 2, Mw = 1.56 × MwYtvA) and a spherical monomer (peak 3, Mw = 1.12 × MwYtvA). Peak 2 (elongated monomer) represents in all YtvA preparations observed (nine in total) the predominant protein fraction. Peaks 1 and 3 are more evident in preparations that contain considerable amount of apoprotein (without the flavin chromophore). Nevertheless, even in these cases, the flavin chromophore is present in all three fractions, as proven by detection at 390 nm (data not shown). Again light activation does not result in appreciable changes in the elution profile. For the three protein constructs, the elution profile is not affected by concentration, in the low range employed here (0.05–2.5 µM).
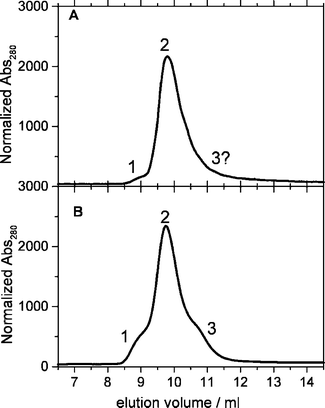 | ||
| Fig. 2 Elution profile of 2 different YtvA preparations. Peak 1 corresponds to Mw = 2.32 × MwYtvA (MwYtvA = 31.36 kDa); peak 2 corresponds to Mw = 1.56 × MwYtvA; peak 3 corresponds to Mw = 1.12 × MwYtvA. A. YtvA : FMN = 1 : 1 (no apoprotein ). B. YtvA : FMN ≈ 2 : 1 (apoprotein is present). | ||
As a whole, the gel filtration experiments show that the LOV domain of YtvA has a strong tendency to dimerize in solution that is not hindered by the N-terminal cap. Dimerization is instead prevented by the presence of the C-terminal domain of the protein, pointing to the fact that the LOV core employs the same surface (partially or totally) for homodimerization and for interdomain interactions.
Circular dichroism spectroscopy
The UV-CD spectra for the three analyzed constructs of YtvA, in the dark adapted state, are shown in Fig. 3. The mean residue ellipticity, ΘMRW, was calculated as explained in the experimental section. The spectra shown are an average of all measurements (5 sets of measurements with 2 different preparations for YLOV; 4 sets of measurements with 2 different preparations for N-YLOV; 11 sets of measurements, 9 different preparations for YtvA), but each single spectral output was analyzed separately using the CCA algorithm. | ||
| Fig. 3 CD spectra in the UV region for (A) the YLOV (dashed line) and N-YLOV (full line) domains and (B) full-length YtvA, in the dark adapted state. | ||
A critical step during CCA analysis of CD data, is the assignment of the component curves (Fig. 4) to specific secondary structures. In the literature there is a large agreement about the CD spectrum of regular α-helices and unordered polypeptides (random coil, RC) that can be assigned to curve I and II respectively (ref. 58 and 59and references therein).
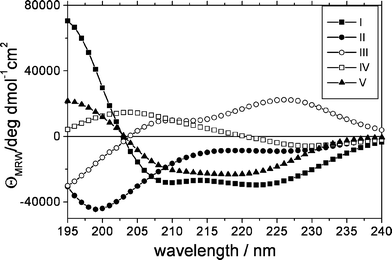 | ||
| Fig. 4 The 5 component curves as extracted from the CCA analysis. The assignment is as follows: I, α-helix, II, unordered structures, III, turns and other structures, IV, distorted/twisted β-strands/parallel β-sheet, V, antiparallel β-sheet (see text for details). | ||
Curve V was assigned to the turn fraction60 and the β-structures were assigned to curve IV + V, whose sum is similar to the curve corresponding to β-strands in Matsuo et al.60 Based on the fact that distorted/twisted β-sheets present a strong positive band in the 190–220 region,61 curve IV is assumed to include this fraction. Furthermore, the antiparallel β-sheet has three allowed transitions (the ππ* transition is splitted),62,63 whereas the parallel β-sheet has two; therefore, we assigned curve V to the regular antiparallel β-sheet and curve IV to twisted + parallel β-sheet. Curve IV may also receive contributions from turn structures, in that different type of turns have very different CD spectra.44,64,65 The results of the CCA analysis, with the curve assignment as discussed above, are reported in Table 1, both for the dark and light-adapted state.
| Secondary structure | LOVa (122 aa)b (%) | N-LOVa (147 aa)b (%) | YtvAa (281 aa)b (%) | |||
|---|---|---|---|---|---|---|
| Dark | Light | Dark | Light | Dark | Light | |
| a The statistical error is the standard deviation and comes from 5 sets of measurements on 2 different preparations for LOV, 4 sets of measurements on 2 different preparations for N-LOV, 11 sets of measurements, and 9 different preparations for YtvA. b The number of aa is given in parentheses, below the percentage, together with the statistical error. c Average squared error, where yi = experimental curve, f(λ) = fitting curve. | ||||||
| I, α-Helix | 16.9 ± 2.7 | 19.5 ± 5.8 | 24.9 ± 2.4 | 27.3 ± 0.9 | 30.9 ± 4.7 | 32.0 ± 6.5 |
| (21 ± 3) | (24 ± 7) | (37 ± 4) | (40 ± 1) | (87 ± 13) | (90 ± 18) | |
| II, RC | 23.9 ± 4.1 | 22.8 ± 3.9 | 24.9 ± 0.9 | 23.6 ± 0.9 | 22.4 ± 1.0 | 21.7 ± 3.5 |
| (29 ± 5) | (28 ± 5) | (37 ± 1) | (35 ± 1) | (63 ± 3) | (61 ± 10) | |
| III, β-Turns/others | 27.9 ± 1.1 | 28.8 ± 2.3 | 27.5 ± 1.0 | 28.3 ± 1.5 | 16.8 ± 3.6 | 18.7 ± 3.5 |
| (34 ± 1) | (35 ± 3) | (40 ± 1) | (42 ± 2) | (47 ± 10) | (52 ± 10) | |
| IV, β-Twisted/β-parallel | 14.7 ± 1.6 | 18.6 ± 1.0 | 12.1 ± 1.6 | 15.9 ± 0.9 | 9.9 ± 4.1 | 11.0 ± 3.7 |
| (18 ± 2) | (23 ± 1) | (18 ± 2) | (23 ± 1) | (28 ± 11) | (31 ± 10) | |
| V, β-Antiparallel | 16.2 ± 4.1 | 10.2 ± 4.7 | 10.6 ± 3.4 | 4.9 ± 2.1 | 19.9 ± 5.6 | 16.6 ± 5.8 |
| (20 ± 5) | (12 ± 6) | (15 ± 5) | (7 ± 3) | (56 ± 16) | (47 ± 16) | |
 c
c
|
1.9 ± 0.4 | 3.6 ± 2.9 | 2.2 ± 1.8 | 2.1 ± 0.7 | 6.2 ± 2.8 | 5.2 ± 2.3 |
To test the quality/reliability of our component assignment, we made a prediction of secondary structure composition based on the three dimensional models of the LOV and STAS domains19,39 and on the consensus method for the remaining parts of the protein.46 The N-cap and the linker region are predicted to be largely helical, whereas the His-tag (20 aa in length) is, as expected, predicted to be unordered (Table 2).
| YtvA segments | Helices | Turns/loops | β-Strands |
|---|---|---|---|
| Number of aa | Number of aa | Number of aa | |
| a Consensus secondary structure prediction at the Pôle Bioinformatique Lyonnais server.46 b Structural homology models of the LOV core19 and of the STAS domain.39 c Predicted number of aa for each of the constructs analyzed. | |||
| His-Taga | — | 20 | — |
| N-Cap1–24a | 10 | 13 | 1 |
| LOV25–126b | 24 | 36 | 42 |
| Linker127–146a | 18 | 2 | — |
| STAS147–254b | 36 | 41 | 31 |
| C-End255–261a | 2 | 4 | 1 |
| YtvAc | 90 | 116 | 75 |
| N-LOVc | 34 | 69 | 43 |
| LOVc | 24 | 56 | 42 |
| CCA analysis—dark state | α-Helix | RC/turns/others | β-Strands |
| YtvA | 87 ± 13 | 110 ± 9 | 84 ± 17 |
| N-LOV | 37 ± 4 | 77 ± 1 | 33 ± 5 |
| LOV | 21 ± 3 | 63 ± 4 | 38 ± 5 |
The comparison with CD data is very good in the case of the helical fraction, although for YtvA the statistical error associated with this component is quite large. This may be due to the variability in the preparations, and/or to the fact that component I and V are in some cases difficult to separate (see Fig. 4 and Table 1). The results confirm that the N-cap and the linker region are mostly helical. For full-length YtvA also the turn/loops and β-strands predicted fractions match the sum of component II + III and IV + V, respectively, within the experimental error, (Table 2), supporting our curve assignment. In the case of LOV and, particularly, N-LOV, the fraction of β-strands is smaller than expected, to the advantage of the RC/turns/others component. Furthermore, LOV domains do not contain parallel β-sheets19,22,23 and the large percentage associated to component IV has to be assigned to the distortion/twisting of the central, antiparallel β-scaffold. This is in contrast with YtvA, for which the number of aa associated to component IV, can be readily explained with the presence of four parallel β-strands localized on the STAS domain and a modest distortion of the overall β-fraction.39 These observations suggest that dimerization in YLOV and N-YLOV markedly affects the central β-sheet of the LOV core (see the docking section).
Inspection of Table 1 and of the light – dark difference spectra (Fig. 5) shows that light activation of the three analyzed constructs does not result in large secondary structure conformational changes, as previously noticed for full-length YtvA.41 A further distortion of the central β-sheet is induced in YLOV and N-YLOV, and the difference spectra are very similar for the two proteins.
 | ||
| Fig. 5 Light – dark plots of the mean residue ellipticity ΘMRW for YtvA (squares + line), LOV (circles + line) and N-LOV (full line), calculated from the average CD spectra (see Fig. 1 and Table 1). | ||
Light-induced changes of the central β-sheet have been recently demonstrated with low temperature Fourier transformed infra-red spectroscopy (FTIR) also for phy3-LOV2.66 The CD light-difference spectrum of phot1-LOV2 was interpreted as a loss of helical structure, but without the support of a detailed data analysis.67
In full-length YtvA there is still a perturbation of the β-fraction, but a distinct change in the turn fraction (positive shoulder at ca. 230 nm in Fig. 5), missing in YLOV and N-YLOV. The determinations are affected by a large error, but confirmed by previous data as obtained with FTIR, that show a distinct difference between YtvA and YLOV in the light-induced changes of the turns fraction (around 1700 cm–1).68 These results could be interpreted as a conformational change transmitted from the LOV core to the STAS domain, but actually we have no hint to localize precisely the position of the altered turn fraction, that could even be on the LOV domain itself and its changes being not detectable in the LOV dimers. Temperature-dependent FTIR experiments show indeed that changes in the turn fraction occur before the conformational alterations of the β-sheet in phy3-LOV2, the latter changes only detectable at room temperature.66 In this view, the light-induced conformational changes could reach the β-scaffold only in the YLOV and N-YLOV dimers and be limited by the presence of the second domain, so that the changes on the turn fraction can persist during the lifetime of the adduct in YtvA.
The YLOV–YLOV dimer
The ClusPro best ranked model (ZDOCK generated) is shown in Fig. 6. A very similar model is ranked at the first position by using the DOT docking software (not shown). | ||
| Fig. 6 (A) The YLOV dimer model (see text for details). (B) The phy3-LOV2 dimer in the crystal unit cell (PDB accession code 1G28, chains a,c).22 (C) Residues at the dimer interface (within 4 Å, shadowed), mapped on the sequence of YLOV and phy3-LOV2. For comparison the interaction hot-spots predicted by PPI-Pred56 (in bold) and by Robetta57 are also shown (in bold and underlined). Arrows indicate the residues interacting with FMN in phy3-LOV2. Secondary structure elements are shown below the phy3-LOV2/YLOV alignment and indicated with conventional letters. E = strands, H = helices, C = unordered. The nomenclature of the secondary structure elements is after Harper et al.24 | ||
The evaluation of the complexes performed with the VADAR tool,50 reveals that this model has a quite large buried surface area as well as a high percentage of buried hydrophobic side-chains (33.76%) (see electronic supplementary information, ESI† ). This feature would favour dimerization in an aqueous environment, and agrees with the fact that YLOV is a stable dimer in solution, even at very low concentrations. The quality of the model, evaluated at the SAVS server (http://nihserver.mbi.ucla.edu/SAVS/) was as an overall good, with 84.1% of residues in most favoured regions, 12.5% of residues in additionally allowed region, 2.3% of residues in generously allowed regions and only 1.1% of residues in disallowed regions of the Ramachandran plot. The Verify-3D54 score (96.1) and the Errat-quality factor53 (98.9) were very high, both indicative of a reasonable and good resolved model-structure. Finally, the dimerization surface predicted with PPI-Pred56 and Robetta,57 identified high scoring regions for YLOV–YLOV interactions within Aβ, Bβ, Hβ and Iβ strands and the Hβ–Iβ loop (Fig. 6).
In the dimer models of Fig. 6, the two YLOV domains face each other with the central β-sheet, presenting an antiparallel mirror symmetry. The interface is mostly stabilized by hydrophobic interactions. This feature is not in common with phy3-LOV2, where a bunch of charged/polar amino acids forms an extended HB (hydrogen bonds) network with the corresponding residues on the second monomer, centered around His1011, Gln1013 (Hβ) and Asp1017 (Hβ–Iβ loop). Interestingly, His1011 and Gln1013 of LOV2 domains (Thr and A/T respectively on LOV1 domains), and this feature may account for the fact that LOV1 has a stronger tendency to dimerize than LOV2 in an aqueous environment.30 Although the dimerization of phy3-LOV2 and the specific orientation of the two monomers may be an artifact of crystallization, a complex very similar to the phy3-LOV dimer is readily obtained by the docking algorithm (not shown) and the residues at the interface are part of the hot spots predicted by PPI-Pred and Robetta (Fig. 6). We note that for YLOV, complexes with similar orientation as phy3-LOV are also detected by the ClusPro docking algorithm (cluster 2 and 8 in the DOT output and cluster 10 in the ZDOCK output, see ESI† ). Our choice of the model in Fig. 6 (cluster 1 for both DOT and ZDOCK outputs) is based on the ClusPro ranking, on the high surface complementarity and interactions symmetry, and on the presence of a cluster of hydrophobic amino acids at the interface, that nicely accounts for the stability of the dimer in solution.
The antiparallel mirror symmetry and the interface observed in our YLOV–YLOV model and in the phy3-LOV dimer is very similar to the one reported for homo and heterodimers of the ARNT PAS-B domain in solution69 and in dimers of the heme-binding PAS domain of E. coli Dos (EcDos)70 and R. meliloti FIXL (RmFIXL).71 An antiparallel mirror symmetry has also been suggested for the LOV–LOV dimer of the FKF1 protein and for phot-LOV1 on the basis of small-angle X-ray scattering experiments, although in that case the authors favoured a different model for the complex, where the two LOV domains do not face each other via the central β-sheet.32,33
The model as in Fig. 6 not only corresponds to the best quality/validation parameters, but also agrees with the observations that in YLOV the β-scaffold is distorted/twisted within the dimer, as indicated by the CD data. With this respect we have to remind that the docking simulation requires that the partners within the complex are kept rigid, a feature that may not be verified in the real complex, as again suggested by the distortion of the β-scaffold. Therefore, the model structure reported in Fig. 6 has to be taken with care and, albeit probably qualitatively correct, may not match the solution dimer-structure as far as the details are concerned.
In the structure of EcDos and RmFIXL PAS domains, the dimers are further stabilized by the helical N-cap and the dimers retain an elongated shape.70,71 In the case of N-YLOV, although the N-cap is helical (from CD data), the dimer is instead approximately spherical (gel filtration ). This observation, together with low similarity to the corresponding sequences in EcDos and RmFIXL, does not allow to build a reliable model of the N-cap in YLOV and of its orientation with respect to LOV core.
Similar considerations apply to a structural model of the full-length protein. The LOV-STAS linker region is predicted to be helical and CD data confirm the prediction (see above), nevertheless we cannot safely state that it assumes an orientation similar to that in phot1-LOV2, namely underneath the central β-scaffold of the LOV core,24 because of low sequence similarity. The amino acid sequence of YtvA Jα-linker is much more similar to the C-terminal extension of the heme binding PAS domain of FIXL from B. japonicum, actually protruding outside the PAS core.72 Although this might be, in the latter case, an artifact of crystallization (in the absence of the associated kinase domain), such orientation of the Jα-linker cannot be excluded. This would imply a direct interaction of the STAS domain with the LOV core, competing with the dimerization surface, different to the structural features proposed for phot as a basis for the self-phosphorylation reaction.24 We must also consider that alternative complex conformations may exist: the linker is not needed for the activation of the kinase activity in phot2 towards a substrate, a reaction carried out via direct interaction between the separately expressed LOV2 and kinase domains.15 As a whole we still have too little structural and functional information about the N-cap and Jα-linker to build a reliable model of full-length YtvA. In order to gain further structural information, e.g., orientation of the helical linker with respect to the LOV core and its relevance in the LOV-STAS interaction, we are designing separated constructs for the STAS domain and the LOV core furnished with the linker region.
We wish to point out that with CD experiments we can only see modifications in the secondary structure elements, but protein movements could occur without large conformational changes of the secondary structure. Furthermore our experiments are not time resolved, therefore we cannot detect transient structural changes occurring within the time-scale for the formation of the adduct (ca. 2 µs).19Another problem is represented by the fact that we are working with a system that only partially resembles physiological conditions. In fact, from a very recent paper we know that YtvA functions within a large macromolecular complex, about which we presently do not have any structural information.37 Some hints about the way the STAS domain is activated may come from our recent experiments showing that YtvA binds Nucleotide TriPhosphate (NTP = GTP, ATP)39 and that light-induced conformational changes are transmitted from the LOV-core to the NTP binding cavity on the STAS domain.39These changes are very small and certainly do not imply large structural changes in YtvA, but may have a larger significance within the macromolecular complex mentioned above.
Conclusions
In this work we have investigated the conformation of YtvA in solution. The analysis of CD spectra by means of the CCA algorithm and curve assignment has been improved with respect to previous work and can be now reliably employed to determine the secondary structural composition of LOV proteins. The LOV domain of YtvA has been proven to be an elongated dimer, stabilized by interactions that involve the β-scaffold, for which we have modelled a structure that agrees with experimental data and bioinformatic analysis. In the N-YLOV construct, the helical N-terminal cap is expected to participate in dimerization, although we could not model the complex due to lack of information about the orientation of this segment. In the full-length protein YtvA, dimerization appears only in case that apoprotein is present, most probably with the formation of heterodimers (apoprotein /FMN-bound YtvA). The data strongly suggest that the β-scaffold is involved both in YLOV dimerization and intraprotein interactions with the linker and/or STAS domain, confirming that this region is a good candidate as a surface responsible for signal transmission to the effector domains in LOV proteins. This latter aspect highlights a sharp similarity with phot-LOV2,24 but the missing light-driven unfolding of the Jα-linker also points to a distinct difference between phot and YtvA, in the way the effector domain is activated. Furthermore, the data presented here suggest that dimerization of LOV domains might play an important regulative role by competing with domain–domain interactions and should be thoroughly investigated.References
- M. H. Hefti, K. J. Francoijs, S. C. de Vries, R. Dixon and J. Vervoort, The PAS fold: A redefinition of the PAS domain based upon structural prediction, FEBS J., 2004, 271, 1198–1208 Search PubMed.
- W. R. Briggs, C. F. Beck, A. R. Cashmore, J. M. Christie, J. Hughes, J. A. Jarillo, T. Kagawa, H. Kanegae, E. Liscum, A. Nagatani, K. Okada, M. Salomon, W. Rüdiger, T. Sakai, M. Takano, M. Wada and J. C. Watson, The phototropin family of photoreceptors, Plant Cell, 2001, 13, 993–997 CrossRef CAS.
- W. R. Briggs and J. M. Christie, Phototropins 1 and 2: versatile plant blue-light receptors, Trends Plant Sci., 2002, 7, 204–210 CrossRef CAS.
- R. Banerjee and A. Batschauer, Plant blue-light receptors, Planta, 2005, 220, 498–502 CrossRef CAS.
- E. Huala, P. W. Oeller, E. Liscum, I. S. Han, E. Larsen and W. R. Briggs, Arabidopsis NPH1: A Protein Kinase with a Putative Redox-Sensing Domain, Science, 1997, 278, 2120–2123 CrossRef CAS.
- E. Knieb, M. Salomon and W. Rüdiger, Autophosphorylation, electrophoretic mobility and immunoreaction of oat Phototropin 1 Under UV and Blue Light, Photochem. Photobiol., 2005, 81, 177–182 CrossRef CAS.
- M. Salomon, J. M. Christie, E. Knieb, U. Lempert and W. R. Briggs, Photochemical and mutational analysis of the FMN-binding domains of the plant blue light receptor phototropin, Biochemistry, 2000, 39, 9401–9410 CrossRef CAS.
- M. Salomon, W. Eisenreich, H. Dürr, E. Scleicher, E. Knieb, V. Massey, W. Rüdiger, F. Müller, A. Bacher and G. Richter, An optomechanical transducer in the blue light receptor phototropin from Avena sativa, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12357–12361 CrossRef CAS.
- M. Kasahara, T. E. Swartz, M. A. Olney, A. Onodera, N. Mochizuki, H. Fukuzawa, E. Asamizu, S. Tabata, H. Kanegae, M. Takano, J. M. Christie, A. Nagatani and W. R. Briggs, Photochemical properties of the flavin mononucleotide-binding domains of the phototropins from Arabidopsis, rice, and Chlamydomonas reinhardtii, Plant Physiol., 2002, 129, 762–773 CrossRef CAS.
- S. Crosson and K. Moffat, Photoexcited structure of a plant photoreceptor domain reveals a light-driven molecular switch, Plant Cell, 2002, 14, 1067–1075 CrossRef CAS.
- C. W. M. Kay, E. Schleicher, A. Kuppig, H. Hofner, W. Rüdiger, M. Schleicher, M. Fischer, A. Bacher, S. Weber and G. Richter, Blue light perception in plants. Detection and characterization of a light-induced neutral flavin radical in a C450A mutant of phototropin, J. Biol. Chem., 2003, 278, 10973–10982 CrossRef CAS.
- J. T. M. Kennis, S. Crosson, M. Gauden, I. H. M. van Stokkum, K. Moffat and R. van Grondelle, Primary Reactions of the LOV2 Domain of Phototropin, a Plant Blue-Light Photoreceptor, Biochemistry, 2003, 42, 3385–3392 CrossRef CAS.
- T. E. Swartz, S. B. Corchnoy, J. M. Christie, J. W. Lewis, I. Szundi, W. R. Briggs and R. A. Bogomolni, The photocycle of a flavin-binding domain of the blue light photoreceptor phototropin, J. Biol. Chem., 2001, 276, 36493–36500 CrossRef CAS.
- T. Kottke, J. Heberle Dominic Hehn and P. Hegemann, Phot-LOV1: Photocycle of a Blue-Light Receptor Domain from the Green Alga Chlamydomonas reinhardtii, Biophys. J., 2003, 84, 1192–1201 CrossRef CAS.
- D. Matsuoka and S. Tokutomi, Blue light-regulated molecular switch of Ser/Thr kinase in phototropin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13337–13342 CrossRef CAS.
- J. M. Christie, T. E. Swartz, R. A. Bogomolni and W. R. Briggs, Phototropin LOV domains exhibit distinct roles in regulating photoreceptor function, Plant J., 2002, 32, 205–219 CrossRef CAS.
- S. Crosson, S. Rajagopal and K. Moffat, The LOV domain family: photoresponsive signaling modules coupled to diverse output domains, Biochemistry, 2003, 42, 2–10 CrossRef CAS.
- A. Losi, The bacterial counterparts of plants phototropins, Photochem. Photobiol. Sci., 2004, 3, 566–574 RSC.
- A. Losi, E. Polverini, B. Quest and W. Gärtner, First evidence for phototropin-related blue-light receptors in prokaryotes, Biophys. J., 2002, 82, 2627–2634 CrossRef CAS.
- U. Krauss, A. Losi, W. Gärtner, K.-E. Jaeger and T. Eggert, Initial characterization of a blue-light sensing, phototropin-related protein from Pseudomonas putida: a paradigm for an extended LOV construct, Phys. Chem. Chem. Phys., 2005, 7, 2229–2236 RSC.
- E. Schleicher, R. M. Kowalczyk, C. W. M. Kay, P. Hegemann, A. Bacher, M. Fischer, R. Bittl, G. Richter and S. Weber, On the reaction mechanism of adduct formation in LOV domains of the plant blue-light receptor phototropin, J. Am. Chem. Soc., 2004, 126, 11067–11076 CrossRef CAS.
- S. Crosson and K. Moffat, Structure of a flavin-binding plant photoreceptor domain: insights into light-mediated signal transduction, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2995–3000 CrossRef CAS.
- R. Fedorov, I. Schlichting, E. Hartmann, T. Domratcheva, M. Fuhrmann and P. Hegemann, Crystal structures and molecular mechanism of a light-induced signaling switch: the Phot-LOV1 domain from Chlamydomonas reinhardtii, Biophys. J., 2003, 84, 2492–2501 Search PubMed.
- S. M. Harper, L. C. Neil and K. H. Gardner, Structural basis of a phototropin light switch, Science, 2003, 301, 1541–1544 CrossRef CAS.
- S. M. Harper, J. M. Christie and K. H. Gardner, Disruption of the LOV-Jalpha helix interaction activates phototropin kinase activity, Biochemistry, 2004, 43, 16184–16192 CrossRef CAS.
- A. Losi, in Flavin photochemistry and photobiology, ed. D.-P. Häder and G. Jori, Elsevier, Amsterdam, 4th edn, 2006, ch. 10, pp. 223–276 Search PubMed.
- T. Eitoku, Y. Nakasone, D. Matsuoka, S. Tokutomi and M. Terazima, Conformational dynamics of phototropin 2 LOV2 domain with the linker upon photoexcitation, J. Am. Chem. Soc., 2005, 127, 13238–13244 CrossRef CAS.
- B. L. Taylor and I. B. Zhulin, PAS domains: internal sensors of oxygen, redox potential and light, Microbiol. Mol. Biol. Rev., 1999, 63, 479–506 CAS.
- M. A. Gilles-Gonzalez and G. Gonzalez, Heme-based sensors: defining characteristics, recent developments, and regulatory hypotheses, J. Inorg. Biochem., 2005, 99, 1–22 CrossRef CAS.
- M. Salomon, U. Lempert and W. Rüdiger, Dimerization of the plant photoreceptor phototropin is probably mediated by the LOV1 domain, FEBS Lett., 2004, 572, 8–10 CrossRef CAS.
- Y. Nakasone, T. Eitoku, D. Matsuoka, S. Tokutomi and M. Terazima, Kinetic Measurement of Transient Dimerization and Dissociation Reactions of Arabidopsis Phototropin 1 LOV2 Domain, Biophys. J., 2006, 91, 645–653 CrossRef CAS.
- M. Nakasako, D. Matsuoka, K. Zikihara and S. Tokutomi, Quaternary structure of LOV-domain containing polypeptide of Arabidopsis FKF1 protein, FEBS Lett., 2005, 579, 1067–1071 CrossRef CAS.
- M. Nakasako, T. Iwata, D. Matsuoka and S. Tokutomi, Light-Induced Structural Changes of LOV Domain-Containing Polypeptides from Arabidopsis Phototropin 1 and 2 Studied by Small-Angle X-ray Scattering, Biochemistry, 2004, 43, 14881–1489 CrossRef CAS.
- P. Ballario, C. Talora, D. Galli, H. Linden and G. Macino, Roles in dimerization and blue light photoresponse of the PAS and LOV domains of Neurospora crassa white collar proteins, Mol. Microbiol., 1998, 29, 719–729 CrossRef CAS.
- L. Aravind and E. V. Koonin, The STAS domain a link between anion transporters and antisigma-factor antagonists, Curr. Biol., 2000, 10, R53–R55 CrossRef CAS.
- S. Akbar, T. A. Gaidenko, K. Min, M. O'Reilly, K. M. Devine and C. W. Price, New family of regulators in the environmental signaling pathway which activates the general stress transcription factor of Bacillus subtilis, J. Bacteriol., 2001, 183, 1329–1338 CrossRef CAS.
- T. A. Gaidenko, T. J. Kim, A. L. Weigel, M. S. Brody and C. W. Price, The blue-light receptor YtvA acts in the environmental stress signaling pathway of Bacillus subtilis, J. Bacteriol., 2006, 188, 6387–6395 CrossRef CAS.
- M. Avila-Perez, K. J. Hellingwerf and R. Kort, Blue light activates the sigmaB-dependent stress response of Bacillus subtilisvia YtvA, J. Bacteriol., 2006, 188, 6411–6414 CrossRef CAS.
- V. Buttani, A. Losi, E. Polverini and W. Gärtner, Blue news: NTP binding properties of the blue-light sensitive YtvA protein from Bacillus subtilis, FEBS Lett., 2006, 580, 3818–3822 CrossRef CAS.
- S. M. Najafi, D. A. Harris and M. D. Yudkin, The SpoIIAA protein of Bacillus subtilis has GTP-binding properties, J. Bacteriol., 1996, 178, 6632–6634 CAS.
- A. Losi, E. Ghiraldelli, S. Jansen and W. Gärtner, Mutational effects on protein structural changes and interdomain interactions in the blue-light sensing LOV protein YtvA, Photochem. Photobiol., 2005, 81, 1145–1152 CrossRef CAS.
- A. Losi, B. Quest and W. Gärtner, Listening to the blue: the time-resolved thermodynamics of the bacterial blue-light receptor YtvA and its isolated LOV domain, Photochem. Photobiol. Sci., 2003, 2, 759–766 RSC.
- A. Perczel, M. Hollosi, G. Tusnady and G. D. Fasman, Convex constraint analysis: a natural deconvolution of circular dichroism curves of proteins, Protein Eng., 1991, 4, 669–679 CAS.
- A. Perczel, K. Park and G. D. Fasman, Analysis of the circular dichroism spectrum of proteins using the convex constraint algorithm: A practical guide, Anal. Biochem., 1992, 203, 83–93 CAS.
- I. R. Bates, P. Matharu, N. Ishiyama, D. Rochon, D. D. Wood, E. Polverini, M. A. Moscarello, N. J. Viner and G. Harauz, Characterization of a Recombinant Murine 18.5-kDa Myelin Basic Protein, Protein Expression Purif., 2000, 20, 285–299 CrossRef CAS.
- C. Combet, C. Blanchet, C. Geourjon and G. Deleage, NPS@: Network Protein Sequence Analysis, Trends Biochem. Sci., 2000, 25, 147–150 CrossRef CAS.
- S. R. Comeau, D. W. Gatchell, S. Vajda and C. J. Camacho, ClusPro: An automated docking and discrimination method for the prediction of protein complexes, Bioinformatics, 2004, 20, 45–50 CrossRef CAS.
- J. G. Mandell, V. A. Roberts, M. E. Pique, V. Kotlovyi, J. C. Mitchell, E. Nelson, I. Tsigelny and L. F. Ten Eyck, Protein docking using continuum electrostatics and geometric fit, Protein Eng., 2001, 14, 105–113 CrossRef CAS.
- R. Chen, L. Li and Z. Weng, ZDOCK: an initial-stage protein-docking algorithm, Proteins, 2003, 52, 80–87 CrossRef CAS.
- L. Willard, A. Ranjan, H. Zhang, H. Monzavi, R. F. Boyko, B. D. Sykes and D. S. Wishart, VADAR: a web server for quantitative evaluation of protein structure quality, Nucleic Acids Res., 2003, 31, 3316–3319 CrossRef CAS.
- R. A. Laskowski, M. W. MacArthur, D. S. Moss and J. M. Thornton, PROCHECK-A program to check the stereochemical quality of protein structures, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef.
- R. W. Hooft, G. Vriend, C. Sander and E. E. Abola, Errors in protein structures, Nature, 1996, 381, 272–272 CrossRef CAS.
- C. Colovos and T. O. Yeates, Verification of protein structures: patterns of nonbonded atomic interactions, Protein Sci., 1993, 2, 1511–1519 CrossRef CAS.
- R. Luthy, J. U. Bowie and D. Eisenberg, Assessment of protein models with three-dimensional profiles, Nature, 1992, 356, 83–85 CrossRef CAS.
- J. Pontius, J. Richelle and S. J. Wodak, Deviations from standard atomic volumes as a quality measure for protein crystal structures, J. Mol. Biol., 1996, 264, 121–136 CrossRef CAS.
- J. R. Bradford and D. R. Westhead, Improved prediction of protein-protein binding sites using a support vector machines approach, Bioinformatics, 2005, 21, 1487–1494 CAS.
- T. Kortemme, D. E. Kim and D. Baker, Computational alanine scanning of protein–protein interfaces, Sci. STKE, 2004, 2004(219), pl2 Search PubMed.
- N. Sreerama, S. Y. Venyaminov and R. W. Woody, Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis, Anal. Biochem., 2000, 287, 243–251 CrossRef CAS.
- N. Sreerama, S. Y. Venyaminov and R. W. Woody, Estimation of the number of alpha-helical and beta-strand segments in proteins using circular dichroism spectroscopy, Protein Sci., 1999, 8, 370–380 CAS.
- K. Matsuo, R. Yonehara and K. Gekko, Improved Estimation of the Secondary Structures of Proteins by Vacuum-Ultraviolet Circular Dichroism Spectroscopy, J. Biochem., 2005, 138, 79–88 CrossRef CAS.
- N. Sreerama and R. W. Woody, Structural composition of betaI- and betaII-proteins, Protein Sci., 2003, 12, 384–388 CrossRef CAS.
- N. J. Greenfield, Analysis of circular dichroism data, Methods Enzymol., 2004, 383, 282–317 CAS.
- R. W. Woody and A. Koslowski, Recent developments in the electronic spectroscopy of amides and alpha-helical polypeptides, Biophys. Chem., 2002, 101–102, 535–551 CrossRef CAS.
- N. J. Greenfield, Methods to estimate the conformation of proteins and polypeptides from circular dichroism data, Anal. Biochem., 1996, 235, 1–10 CrossRef CAS.
- C. T. Chang, C. S. Wu and J. T. Yang, Circular dichroic analysis of protein conformation: inclusion of the beta-turns, Anal. Biochem., 1978, 91, 13–31 CAS.
- T. Iwata, D. Nozaki, S. Tokutomi and H. Kandori, Comparative investigation of the LOV1 and LOV2 domains in Adiantum Phytochrome3, Biochemistry, 2005, 44, 7427–7434 CrossRef CAS.
- S. B. Corchnoy, T. E. Swartz, J. W. Lewis, I. Szundi, W. R. Briggs and R. A. Bogomolni, Intramolecular proton transfers and structural changes during the photocycle of the LOV2 domain of phototropin 1, J. Biol. Chem., 2003, 278, 724–731 CrossRef CAS.
- T. Bednarz, A. Losi, W. Gärtner, P. Hegemann and J. Heberle, Functional variations among LOV domains as revealed by FT-IR difference spectroscopy, Photochem. Photobiol. Sci., 2004, 3, 575–579 RSC.
- P. B. Card, P. J. A. Erbel and K. H. Gardner, Structural Basis of ARNT PAS-B dimerization: use of a common beta-sheet interface for hetero- and homodimerization, J. Mol. Biol., 2005, 353, 664–677 CrossRef CAS.
- H. J. Park, C. Suquet, J. D. Satterlee and C. Kang, Insights into signal transduction involving PAS domain oxygen-sensing heme proteins from the X-ray crystal structure of Escherichia Coli Dos Heme Domain (EcDosH), Biochemistry, 2004, 43, 2738–2746 CrossRef CAS.
- H. Miyatake, M. Mukai, S. Y. Park, S. Adachi, K. Tamura, H. Nakamura, K. Nakamura, T. Tsuchiya, T. Iizuka and Y. Shiro, Sensory mechanism of oxygen sensor FixL from Rhizobium meliloti: crystallographic, mutagenesis and resonance Raman spectroscopic studies, J. Mol. Biol., 2000, 301, 415–431 CrossRef CAS.
- W. Gong, B. Hao, S. S. Mansy, G. Gonzalez, M. A. Gilles-Gonzalez and M. K. Chan, Structure of a biological oxygen sensor: a new mechanism for heme-driven signal transduction, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15177–15182 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Calculation of accessible and buried surface areas using the VADAR tool. See DOI: 10.1039/b610375h |
| This journal is © The Royal Society of Chemistry and Owner Societies 2007 |