Visible light decomposition of ammonia to N2 with Ru(bpy)32+ sensitizer
Junichi
Nemoto
,
Chihiro
Harada
,
Yoshihito
Takei
,
Naoto
Katakura
and
Masao
Kaneko
*
Faculty of Science, Ibaraki University, 2-1-1 Bunkyo, Mito, 310-8512, Japan. E-mail: mkaneko@mx.ibaraki.ac.jp
First published on 24th November 2006
Abstract
Visible light decomposition of aqueous NH3 to N2 was investigated using a photocatalyst aqueous solution based on molecular photoelectron relay systems of either sensitizer (tris(2,2′-bipyridine)ruthenium(II), (Ru(bpy)32+)/potassium peroxodisulfate(K2S2O8) or Ru(bpy)32+/methylviologen dichloride(MV2+)/O2, capable of using visible light instead of UV-driven semiconductors such as TiO2. It was confirmed by using an in situ visible absorption spectral change under irradiation that the Ru(II) complex is oxidized to the Ru(III) complex by K2S2O8, and that the Ru(III) complex formed is stable without NH3, while the added NH3 was oxidized by the Ru(III) complex to produce the Ru(II) complex. In the presence of 1 mM NH3 aqueous solution, the Ru(III) complex was the predominant species under the photostationary state, but in the presence of 100 mM NH3, Ru(II) predominated. Gas-chromatographic analysis of the gaseous phase in the presence of 8.1 M NH3 showed that the photochemical oxidation of ammonia yielded N2. It was also demonstrated by using the in situ visible absorption spectrum under irradiation of the NH3 (1 M)/Ru(bpy)32+ (0.1 mM)/MV2+ (10 mM) system under Ar that MV+˙ is accumulated, showing that NH3 works as an electron donor for MV+˙ accumulation with simultaneous formation of the oxidized product of ammonia ((NH3)ox) without producing N2. It was suggested that the reduced product (MV+˙) and the oxidized product ((NH3)ox) are in a kind of dynamic equilibrium prohibiting further oxidation of (NH3)ox by Ru(bpy)33+ to N2. In the O2 atmosphere, the oxidation of MV+˙ to MV2+ takes place to accumulate Ru(III) complex, so that (NH3)ox was further oxidized to N2. The high activity of IrO2 as a cocatalyst in this system was demonstrated.
Introduction
Our ecosystem discharges a large amount of nitrogen compounds, which belong to the four major categories of global pollutants.1 As for such compounds of nitrogen, there are nitric oxide (NO), nitrogen dioxide (NO2), ammonia (NH3), nitrous oxide (N2O), peroxy acetyl nitrate, nitric acid (HNO3), ammonium (NH4+), etc. The largest emission of nitrogen compounds in our society is NH3, the amount of which is larger than NOx (including NO and NO2).2 In the ground NH3 is rapidly converted to NH4+ because of the abundance of hydrogen ions. The bacteria, including Nitrobacter and Nitrosomonas, contribute to the conversion of NH4+ to NO3−, which is referred to as nitrification.3 Under anaerobic conditions, NO3− is converted to N2 gas by some bacteria,4,5 which is defined as denitrification. In total, these processes are known as the nitrogen cycle,6,7 which has kept our environment safe with regard to nitrogen problems.8,9 However, our society is now releasing too much NH3 especially from livestock wastes, which far exceeds the amount that is decomposed into safe N2 by the natural nitrogen cycle. It is an important and urgent issue to decompose NH3 into N2 to protect our environment against pollution.Generally, treatments of waste water are biological, chemical and electrochemical. Treatments other than the electrochemical method require a higher construction cost, a longer time, an additive process, and heat energy, etc. The electrochemical processes can completely convert organic contaminants into gases such as N2, CO2, and H2, etc.8 However, a photochemical process using visible light from sunshine is more favorable for solving these problems because of its economical and environmental compatibility with nature. Since nitrogen compounds in livestock wastes are converted finally to ammonia by enzymes such as urease, if ammonia can be converted to N2, it can solve the livestock waste problem with regard to nitrogen.
Many organic and inorganic compounds have been decomposed by a photocatalyst such as TiO2.10–15 The photodecomposition of ammonia in neutral water has also been reported by using TiO2-supported Pt (Pt–TiO2) or Pd (Pd–TiO2) catalyst, but only biohazardous nitrogen oxides were obtained.16 Photodecomposition of NH3 to produce N2 has also been demonstrated by using platinized TiO2.17,18 We found recently that Pt–TiO2 could photodecompose aqueous ammonia into N2 and H2 with a stoichiometric 1 : 3 ratio (volume) if reacted under alkaline conditions. However, visible light conversion of NH3 to N2 has not been reported except in our recent communication.19 This system is composed of either tris(2,2′-bipyridine)ruthenium(II) (Ru(bpy)32+)/potassium peroxodisulfate (K2S2O8) or Ru(bpy)32+/methylviologen dichloride (MV2+)/O2.20 Since ∼50% of the solar spectrum is visible light composed of the wavelength of 400–760 nm, it is more beneficial to use visible light for decomposing NH3 to N2. In the present paper, we will report the details of visible light decomposition of ammonia into N2 as a candidate for a future artificial solar nitrogen cycle system.
Experimental
Materials
Tris(2,2′-bipyridine)ruthenium(II) (Ru(bpy)32+) and iridium(IV) oxide (IrO2) were purchased from Aldrich Co. Ltd., and methylviologen dichloride (MV2+) from Tokyo-Kasei Co. Ltd. Pt-black and an ammonia aqueous solution (30%) were obtained from Kanto Chemical Co., Inc. Potassium peroxodisulfate (K2S2O8) was from Wako Pure Chemical Ind. Ltd. and Ar gas with >99% purity was obtained from Nippon Sanso Corp. All the chemicals were of the purest grade available and used as received. Ru(bpy)3(ClO4)2 was synthesized as follows: Ru(bpy)3Cl2 (4.7 × 10−4 mol per 50 ml aqueous solution) and NaClO4 (8.5 × 10−3 mol per 50 ml aqueous solution) were mixed, filtered, washed with pure water and dried in a vacuum.In situ measurement of the visible absorption spectrum under photoirradiation
A diode array spectrophotometer, Shimadzu Multispec-1500, was used for the in situ spectroscopy under photoirradiation conditions.21 In this instrument the mechanical shutter speed to take in the monitoring light onto the diode array is about 0.1 s thus allowing the minimum time interval between measurements to be 0.1 s. A cell box was designed and fabricated to minimize the effects of scattering excitation light. The spot shape of the monitoring light (light intensity 0.54 mW cm−2) on the solution surface was an ellipse (the line of upsides 8 mm and the minor axis 5 mm). It was confirmed that the excitation light does not affect the photochemical process in sample solutions. A quartz cell with a 1 cm light pass length and a 5 cm height was used. A 3 ml aqueous solution containing 0.1 mM Ru(bpy)32+, 10 mM K2S2O8, and NH3 was used, deaerated by bubbling argon gas for 10 min, and all the measurements were carried out at 25 °C. The solution surface (area, 1 cm × 3 cm) was irradiated from the horizontal direction by the excitation light from a 500 W xenon lamp through a UV cut-off (L-42) and IR cut-off filters (IRA-25S) (light intensity 101 mW cm−2). Visible light from a 100 W halogen lamp without filters (light intensity 167 mW cm−2) was irradiated for gaseous product analysis. In some cases the visible absorption of a solution before irradiation was taken as a base line, and the net change of the absorbance due to photoreaction was recorded.Photodecomposition of an NH3 aqueous solution using Ru(bpy)32+, MV2+ and O2
A 5 ml aqueous solution containing 0.1 mM Ru(bpy)32+, 10 mM MV2+, and 8.1 M NH3 in a cylindrical reactor (12 ml) was reacted under irradiation with or without 1 mM IrO2. The reactor was sealed with a rubber septum, through which the gas phase could be sampled with a syringe. The photoirradiation was carried out using a 100 W halogen lamp without filters (light intensity 167 mW cm−2) at 25 °C. A sample aliquot of 100 µl gas was taken with a 100 µl syringe for analysis by gas chromatography.Results and discussion
The typical in situ visible absorption spectrum of the solution composed of 0.1 mM Ru(bpy)32+ and 10 mM K2S2O8 in the photoirradiated state is shown in Fig. 1(a). The absorption peak at 452 nm due to Ru(bpy)32+ disappeared quickly on irradiation, reaching the absorption by Ru(bpy)33+ having a peak at 420 nm. The visible absorption of the above solution before irradiation was taken as a base line, and the net change of the solution by photoreaction was monitored and the result is shown in Fig. 1(b). The absorbance change of the peak at 452 nm is shown in Fig. 1(c). When switching on the irradiation, the absorbance at 452 nm decreased steeply and reached saturation within 100 s. After switching off the irradiation, the absorbance increased slightly. This spectral change means that the Ru(bpy)33+ formed was almost stable in the present system. Further irradiation caused a slight decrease of the absorbance showing total recovery of the Ru(III) complex by K2S2O8. | ||
| Fig. 1 In situ visible absorption spectral change of the aqueous solution of 0.1 mM Ru(bpy)32+ and 10 mM K2S2O8(pH not adjusted), under visible light irradiation from a 500 W xenon lamp through a UV cut-off filter (L-42) and IR cut-off filter (IRA-25S) with a light intensity of 101 mW cm−2 under an Ar atmosphere. (a) Typical absorbance of Ru(bpy)32+ and Ru(bpy)33+, (b) spectral change after irradiation with the absorbance before irradiation taken as the base line, (c) time-course of the absorbance change at 452 nm. The spectrum was measured with a 5 s interval in (b). | ||
The spectral changes in the presence of different concentrations of ammonia (1 mM and 100 mM) are shown in Fig. 2(a) and (b), respectively. The different NH3 concentrations greatly affected the absorbance change. In the 1 mM NH3 solution the absorbance change was quick and after switching off the irradiation reached a stable state. It should be noted that the irradiation of the aqueous solution of Ru(bpy)32+ by visible light in the presence of K2S2O8 (10 mM) induced the formation of Ru(bpy)33+ by oxidation of the photoexcited Ru complexes by K2S2O8 in a similar way as in Fig. 1(c). It is shown that in the 1 mM NH3 solution, almost the same reaction occurs except that there is a partial existence of the Ru(II) complex in the photoirradiated state. On the other hand, in the presence of 100 mM NH3, the absorbance change of Ru(bpy)33+ was much slower and more incomplete than that with 1 mM NH3, as shown in Fig. 2(b). The absorbance decreased slowly and after switching off the irradiation the absorbance increased clearly, showing a quick reaction of the Ru(III) complex with NH3. Further irradiation induced the absorbance decrease again. These absorbance changes indicate that the Ru(II) and Ru(III) complexes are in a dynamic equilibrium with NH3.
 | ||
| Fig. 2 In situ visible absorption spectral change of the aqueous solution of 1 mM or 100 mM NH3, 0.1 mM Ru(bpy)32+ and 10 mM K2S2O8, under visible light irradiation from a 500 W xenon lamp through a UV cut-off filter (L-42) and IR cut-off filter (IRA-25S) with a light intensity of 101 mW cm−2 under an Ar atmosphere. (a) 1 mM NH3 pH 5.6, (b) 100 mM NH3 (pH 11.0). The spectrum was measured with a 5 s interval for 5 min. The right figures represent the time dependent absorbance changes at 452 nm over 5 min. | ||
The photochemical reaction product was analysed for the NH3/Ru(bpy)32+/K2S2O8 system by gas chromatography showing N2 formation as reported earlier.20 During the irradiation fine bubbles of N2 in the NH3 (8.1 M)/Ru(bpy)32+ (0.1 mM)/K2S2O8 (10 mM) system were observed and the time course of the N2 evolution is shown in Fig. 3. The N2 evolution increased linearly up to 5 min and then reached a plateau at 298 µl, which is 73% of the theoretical value (407 µl) when the 5 ml aqueous solution of 10 mM K2S2O8 (sacrificial acceptor) is completely consumed in a two-electron reduction process. At this plateau, the decomposition ratio of NH3 is 0.4% of the initial concentration. i.e., this decomposition ratio is limited by the amount of the acceptor K2S2O8 (10 mM). In a separate experiment gas evolution was not observed without Ru(bpy)32+ showing evidently that photodecomposition does not occur in an NH3/K2S2O8 system.
 | ||
| Fig. 3 Time dependent photochemical N2 evolution in the aqueous NH3(8.1M)/Ru(bpy)32+(0.1 mM)/K2S2O8 (10 mM) system at pH 12.9 under an Ar atmosphere. The dashed line indicates the theoretical amount of N2 evolution when K2S2O8 (sacrificial acceptor, 10 mM in 5 ml aqueous solution) is completely consumed. Visible light irradiation from a 100 W halogen lamp with a light intensity of 167 mW cm−2. | ||
To investigate the intermediate of the NH3 photodecomposition into N2, the photochemical decomposition of either NH3 or N2H4 in an aqueous solution of Ru(bpy)3(ClO4)2 (1 mM)/K2S2O8 (0.1 mM) was studied and the result is shown in Table 1. The concentration of the NH3 solution (100 mM) is 10 times as much as that of the N2H4 solution (10 mM), but the N2 evolution by N2H4 in 1 h was five times as much as that by NH3. It means that N2H4 is more easily oxidizable than NH3 in this system. The spectral change in the N2H4 (100 mM)/Ru(bpy)32+(0.1 mM)/K2S2O8 (10 mM) system by taking the absorbance before irradiation as the base line is shown in Fig. 4 where the absorbance change at 452 nm was not clearly observed supporting a very quick decomposition of N2H4 by Ru(bpy)33+ to N2. N2H4 could be a candidate for an intermediate in the NH3 decomposition, but the present results could not confirm conclusively the nature of the intermediate due to the rapid N2H4 reaction. Anyway we can at least say that the reaction occurs by photochemical oxidation of the Ru(II) complex to Ru(III) by K2S2O8, leading to the oxidation of NH3 by the Ru(III) complex to yield N2 (Scheme 1) possibly via short-lived N2H4 under the present conditions.
 | ||
| Fig. 4 In situ visible absorption spectral change of the aqueous solution, 100 mM N2H4, 0.1 mM Ru(bpy)32+ and 10 mM K2S2O8 at pH 11.0, under visible light irradiation from a 500 W xenon lamp through a UV cut-off filter (L-42) and IR cut-off filter (IRA-25S) with a light intensity of 101 mW cm−2 under an Ar atmosphere. The spectrum was measured with a 5 s interval for 5 min by taking the absorbance before irradiation as the base line. | ||
 | ||
| Scheme 1 | ||
Methylviologen (MV2+) is a well-known mediator for proton reduction towards H2 evolution in the presence of a sacrificial donor.11–13 The in situ visible absorption spectrum under irradiation of the NH3 (1 M)/Ru(bpy)32+ (0.1 mM)/MV2+ (10 mM) system under Ar is shown in Fig. 5(a) where the accumulation of the viologen cation radical (MV+˙) having absorption maxima at 395 and 603 nm was clearly observed. It should be noted here that MV+˙ is not accumulated in the two components system, Ru(bpy)32+/MV2+, since the recombination reaction between Ru(bpy)33+ and MV+˙ is very rapid. In the above NH3/Ru(bpy)32+/MV2+ system, we confirmed by another experiment that NH3 did not quench the photoexcited Ru(bpy)32+ complex. It has been well established that electron transfer takes place from the photoexcited Ru(bpy)32+ to MV2+. For these reasons, it could be concluded that in this system methylviologen works as an acceptor from the photoexcited Ru complex to produce MV+˙; ammonia works as an electron donor to the formed Ru(III) complex resulting in the oxidized compound of ammonia ((NH3)ox). The time course of the absorbance change at 606 nm due to MV+˙ for the NH3/Ru(bpy)32+/MV2+ system is shown in Fig. 5(b) where the change of the absorbance increased steeply initially and then became a gentle slope within 1 h. In the 1 h irradiation, N2 evolution was not observed under Ar. This reaction mechanism is represented in Scheme 2 where the NH3 molecule works as the electron donor for the MV2+ reduction, resulting in accumulation of the oxidized product of ammonia ((NH3)ox). As a candidate for the (NH3)ox, hydrazine or NH3+˙ might be possible, but the structure cannot yet be determined. The accumulated MV+˙ disappeared quickly upon switching off the irradiation. It is strongly suggested that the reduced product (MV+˙) and the oxidized product ((NH3)ox) are in a kind of dynamic equilibrium prohibiting further oxidation of (NH3)ox by Ru(bpy)33+ to N2.
 | ||
| Fig. 5 In situ visible absorption spectral change of the aqueous solution, 1 M NH3, 0.1 mM Ru(bpy)32+ and 10 mM MV2+(pH 11.5), under visible light irradiation from a 500 W xenon lamp through a UV cut-off filter (L-42) and IR cut-off filter (IRA-25S) with a light intensity of 101 mW cm−2 under an Ar atmosphere. The spectrum was measured with a 5 s interval by taking the absorbance before irradiation as the base line; total photoreaction time being 1 h in (a). The right figure represents the time dependent absorbance change at 606 nm. | ||
 | ||
| Scheme 2 | ||
As published in our earlier communication,20 under an O2 atmosphere, 860 µl N2 was produced by irradiation of an aqueous solution of NH3 (10 M)/Ru(bpy)32+(0.1 mM)/MV2+(10 mM) after 9 h without MV+˙ accumulation. This mechanism can be represented in Scheme 3. In this system, when O2 was absent (Scheme 2), the predominant species in the reaction mixture are MV+˙, (NH3)ox, and Ru(bpy)32+, because of the absence of the absorbance change around 450 nm due to the Ru(II) complex (see Fig. 5(a)). In the O2 atmosphere, the oxidation of MV+˙ to MV2+ takes place to accumulate Ru(III) complex and (NH3)ox leading to further oxidation to N2.
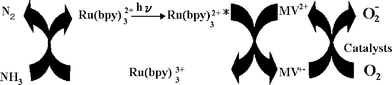 | ||
| Scheme 3 | ||
We investigated the effect of catalysts on the photodecomposition of NH3 by Ru(bpy)32+/MV2+ in an aqueous solution under air and the result is shown in Fig. 6. In this figure, the effect of IrO2 as a catalyst on the photochemical decomposition of NH3 under air is very large. Under these reaction conditions, N2 evolution was not efficient without IrO2. On the contrary, when using IrO2 as a catalyst, NH3 decomposed quickly to N2 and the volume of the evolved N2 gas reached a plateau at 5 h after a few hours induction time and the following steep increase. In Scheme 3 the reduction of MV2+ to MV+˙ by Ru(bpy)32+* and also the oxidation of MV+˙ by O2 are very rapid, so that the oxidation of NH3 by Ru(bpy)33+ must be rate-determining. It is therefore inferred that the IrO2 catalyst would catalyse the oxidation of NH3 to N2 in Scheme 3. The catalyst would collect positive charges to enhance the multi-electron process of the reaction. Other catalysts such as RuO2, Pt black, and RuO2/Pt black co-catalyst, have also been tested, but did not give good results.
 | ||
| Fig. 6 Time dependent photochemical N2 evolution in aqueous NH3 (8.1 M)/IrO2(1 mM)/Ru(bpy)32+ (0.1 mM)/MV2+(10 mM) system (pH not adjusted) (5 ml solution) under air by visible light irradiation from a 100 W halogen lamp without filters with a light intensity of 167 mW cm−2. | ||
The decomposition ratio of NH3 in the experiment shown by Fig. 6 was 0.2% of the theoretical value. This low decomposition ratio of NH3 might be explained by the following. At first, we considered the number of electrons involved in this photodecomposition reaction. It should be noted that N2 evolution is caused by the oxidation of NH3, in which 6 electrons are involved. If the O2 were consumed by two-electron reduction, the amount of O2 consumed should be 120 µmol when the evolved N2 is 40 µmol (after 22 h). This estimation does not coincide with the experimental result in Fig. 6. In contrast, when assuming that O2 was consumed by four-electron reduction, the amount of the O2 consumed should be 60 µmol when the evolved N2 is 40 µmol after 22 h. However, in our experimental result we observed that the O2 consumed was about 40 µmol after 22 h. Therefore, the excess of reduction power (40 µmol × 6–40 µmol × 4 = 80 µmol electrons) must have been utilized in some other reduction process. As for possible candidates, the two-electron irreversible reduction of MV2+ to MV0 is the most probable. If this is the case, the amount of the MV2+reduced irreversibly is calculated to be 40 µmol, which is equivalent to 80% of MV2+ in the cell (50 µmol). It is likely that this irreversible two-electron reduction of MV2+ leads to termination of the oxidation reaction of NH3. When the volume of N2 evolution reached a plateau, the amount of electrons consumed by the NH3 oxidation is 240 µmol (= 40 µmol × 6), so that the turnover number (TN) for the Ru complex (5 × 10−7 mol present in the reaction cell) is calculated to be 480 in 22 h, while for MV2+ (5 × 10−5 mol present in the same cell) it is 4.8 in 22 h. It is concluded that both the Ru complex and the MV2+ work as catalysts.
Thus, in both the NH3/Ru(bpy)32+/K2S2O8 under an Ar atmosphere and NH3/IrO2/Ru(bpy)32+/MV2+ under air, the visible light decomposition of NH3 to N2 in an aqueous solution was investigated by using in situ visible absorption spectra and gas chromatography analysis. These molecule-based photocatalysts can work under visible light, and could be utilized for solving the NH3/N2 circulation problem by an artificial nitrogen cycle converting NH3 to N2 with the help of solar irradiation.
Acknowledgements
The present work has been supported by the Grant-in-Aid for Scientific Research (No. 18550164) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of the Japanese Government.References
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere, Academic Press, New York, 2000 Search PubMed.
- S. V. Krupa, Effect of Atmospheric Ammonia (NH3) on Terrestrial Vegetation: A Review, Environ. Pollut., 2003, 124, 179–221 CrossRef CAS.
- S. J. Ferguson, Nitrogen Cycle Enzymology, Curr. Opin. Chem. Biol., 1998, 2, 182–193 CrossRef CAS.
- H. Bothe, G. Jost, M. Schloter, B. B. Ward and K.-P. Wtzel, Molecular Analysis of Ammonia Oxidation and Denitrification in Natural Environments, FEMS Microbiol. Rev., 2000, 24, 673–690 CrossRef CAS.
- I. Moura and J. J. G. Moura, Structural Aspects of Denitrifying Enzymes, Curr. Opin. Chem. Biol., 2001, 5, 168–175 CrossRef CAS.
- R. W. Ye and S. M. Thomas, Microbial Nitrogen Cyles: Physiology, Genomics and Applications, Curr. Opin. Microbiol., 2001, 4, 307–312 CrossRef CAS.
- D. J. Richadson and N. J. Watmough, Inorganic Nitrogen Metabolism in Bacteria, Curr. Opin. Chem. Biol., 1999, 3, 207–219 CrossRef CAS.
- C. Feng, N. Sugihara, S. Shimada and T. Maekawa, Development of a High Performance Electrochemical Wastewater Treatment System, J. Hazard. Mater., 2003, B103, 65–78 CrossRef.
- P. Juteau, D. Trembly, C.-B. Ould-Moulaye, J.-G. Bisaillon and R. Beaudet, Swine Waste Treatment by Self-heating Aerobic Thermophilic Bioreactors, Water Res., 2004, 38, 539–546 CrossRef CAS.
- A. Fujishima and K. Honda, Electrochemical Photolysis of Water at a Semiconductor Electrode, Nature, 1972, 238, 37–38 CAS.
- A. Fujishima, K. Hashimoto and T. Watanabe, TiO2 Photocatalysis-Fundamentals and Applications, BKC Inc.,Tokyo, 1999 Search PubMed.
- Photosensitization and Photocatalysis Using Inorganic and Organometallic Compounds, ed. K. Kalyanasundaram and M. Graetzel, Kluwer Academic Publishers, Dordrecht, 1993 Search PubMed.
- Photocatalysis-Science and Technology, ed. M. Kaneko and I. Okura, Kodansha/Springer, Tokyo, Berlin, London, 2002 Search PubMed.
- D. Chatterjee and S. Dasgupta, Visible Light Induced Photocatalytic Degradation of Organic Pollutants, J. Photochem. Photobiol., C, 2005, 6, 186–205 CrossRef CAS.
- A. Fujishima, T. N. Rao and D. A. Tryk, Titatnium Dioxide Photolysis, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- J. Taguchi and T. Okuhara, Selective Oxicative Decomposition of Ammonia in Neutral Water to Nitrogen over Titania-supported Platinum or Palladium Catalyst, Appl. Catal., A, 2000, 194–195, 89–97 CrossRef CAS.
- J. Lee, H. Park and W. Choi, Selective Photocatalytic Oxidation of NH3 to N2 on Platinized TiO2 in Water, Environ. Sci. Technol., 2002, 36, 5462–5468 CrossRef CAS.
- W. Choi, J. Lee, S. Kim, S. Hwang, C. Lee and T. K. Lee, Nano Pt Particles on TiO2 and Their Effects on Photocatalytic Reactivity, J. Ind. Eng. Chem., 2003, 9, 96–101 Search PubMed.
- M. Kaneko, N. Gokan, N. Katakura, Y. Takei and M. Hoshino, Artificial Photochemical Nitrogen Cycle to Produce Nitrogen and Hydrogen from Ammonia by Platinized TiO2 and Its Application to a Photofuel Cell, Chem. Commun., 2005, 1625–1627 RSC.
- M. Kaneko, N. Katakura, C. Harada, Y. Takei and M. Hoshino, Visible Light Decomposition of Ammonia to Dinitrogen by a New Visible Light Photocatalytic System Composed of Sensitizer (Ru(bpy)32+, Electron Mediator (Methylviologen) and Electron Acceptor (Dioxygen), Chem. Commun., 2005, 3436–3438 RSC.
- M. Kaneko, K. Suzuki and Y. Kaburagi, Investigation of Photostationary State of Chromophor/acceptor and Related Multistep Electron Relay by in situ Visible Absorption Spectroscopy, J. Photochem. Photobiol., A, 2005, 169, 71–77 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2007 |