Stereoselective synthesis, natural occurrence and CB2 receptor binding affinities of alkylamides from herbal medicines such as Echinacea sp.
N.
Matovic
a,
A.
Matthias
b,
Jürg
Gertsch
c,
Stefan
Raduner
c,
K. M.
Bone
bd,
R. P.
Lehmann
b and
J. J.
DeVoss
*a
aChemistry, School of Molecular and Microbial Sciences, The University of Queensland, Brisbane, 4072, Australia. E-mail: j.devoss@uq.edu.au; Fax: +61 7 3365 4299; Tel: +61 7 3365 3825
bMediHerb Research Laboratories, 3/85 Brandl Street, Eight Mile Plains, 4113, Australia. E-mail: reg@mediherb.com.au; Fax: +61734236599; Tel: +61734236521
cDepartment of Chemistry and Applied Biosciences, ETH Zürich, CH-8093, Zürich, Switzerland. E-mail: juerg.gertsch@pharma.ethz.ch; Fax: +41446331366; Tel: +41446337374
dSchool of Health, University of New England, Armidale, 2351, Australia. Fax: +61746610788; Tel: +61746610700
First published on 29th November 2006
Abstract
A divergent synthesis of (2E,4E,8E,10E)- and (2E,4E,8E,10Z)-N-isobutyldodeca-2,4,8,10-tetraenamides from pent-4-yn-1-ol allowed identification of the (2E,4E,8E,10Z)-isomer for the first time in Echinacea species. A short, stereoselective synthesis of the (2E,4E,8E,10Z)-isomer is also described which allowed further biological evaluation of this material, and the demonstration that this isomer does not occur in Spilanthes mauritiana as previously reported.
Introduction
Polyunsaturated isobutyl alkyl amides are found in a variety of plants that posses interesting biological activities. The two tetraene isomers 1 and 2(Fig. 1), are the major lipophilic constituents found in the two commonly used Echinacea species (E. purpurea and E. angustifolia).1 They have also been found in other medicinal plants such as Salmea scandens,2 and Asarum forbesii Maxim.3S. scandens has various uses in traditional medicine, especially as an anaesthetic for treating toothache,4 while A. forbesii Maxim has been used for its analgesic, antitussive, anti-allergic and diuretic effects.5 The 8E,10Z tetraene isomer 3 has also been reported to occur in Spilanthes mauritiana, and the isolated compound was demonstrated to be a potent mosquito larvicide.6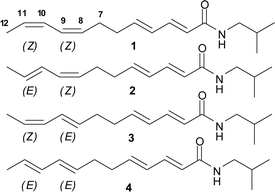 | ||
| Fig. 1 Four geometric isomers about the 8,10 double bonds of isobutyldodeca-2(E), 4(E), 8, 10-tetraenamide are possible. Only 1, 2 and 3 were identified in E. purpurea and E. angustifolia. | ||
E. purpurea and E. angustifolia are the most widely used species in Echinacea derived herbal medicines and together form the basis of the Echinacea industry, on which an estimated $US500 million dollars was spent in the USA alone in 2002.7 These preparations are popularly believed to act via modulating the cellular immune system and frequently taken for prevention or treatment of cold and flu like symptoms.8 The phytochemical profile of E. purpurea and E. angustifolia is well documented, consisting of caffeic acid derivatives, mono and polysaccharides and a complex mixture of alkyl amides. It is still unclear exactly which components are responsible for the reported effects of Echinacea extracts, however recent publications have focused on the alkyl amide constituents found in commercial (mainly ethanolic) extracts. Although the effectiveness of Echinacea in preventing viral illness has recently been called into question,9 it is clear that the alkyl amides do possess significant biological activities. The alkyl amides are the only components of Echinacea extracts found to cross the intestinal barrier,10 and they have been reported to display anti-inflammatory activity.11 Certain alkyl amides have also recently been shown to act as full agonists at the cannabinoid type-2 (CB2) receptor.12
Analysis of the alkyl amides is typically performed using RP-HPLC, but this fails to completely resolve the predominant tetraenes 1 and 2, which are thus reported as a mixture. GCMS analysis not only separates isomers 1 and 2, but also reveals a third component (Fig. 2) with mass spectral fragmentation characteristic of tetraene alkyl amides.13 This third component was suspected to be either 3 or 4, cis/trans isomers of 1 and 2. We identified tetraenes 1 and 2 by comparison to synthetic standards available in our laboratory but identification of the third component required the synthesis of 3 and 4.
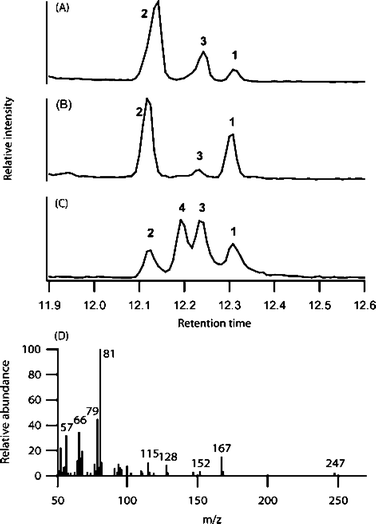 | ||
| Fig. 2 GC/MS trace of E. angustifolia (A) and E. purpurea (B) root extracts and synthetic standards (C). Mass spectrum of 3 (D). | ||
Results and discussion
The simplest approach for rapid access to both 3 and 4 in isomerically pure form was by a divergent synthesis that started from pent-4-yn-1-ol, available in two steps from commercial tetrahydrofurfural alcohol (Scheme 1).14 THP protection, followed by treatment with butyl lithium and then paraformaldehyde gave propargyl alcohol 6, which after treatment with LiAlH4 cleanly provided the E-allyl alcohol 7. Consecutive Swern and Wittig reactions resulted in a 1 : 1 mixture of 8a and 8b that could be separated into the pure isomers by chromatography utilising silver nitrate impregnated silica.15 The stereochemistry of the dienes was readily ascertained by NMR. In the 1H NMR of 8a the two hydrogens belonging to the newly formed Z-double bond were easily distinguished (CDCl3, δ 5.37, dq and δ 5.97, br t), and showed the 11 Hz coupling expected of a cis olefin. The hydrogens of the newly formed E-double bond of 8b could only be clearly identified in C6D6, but displayed the 14 Hz coupling expected of a trans alkene.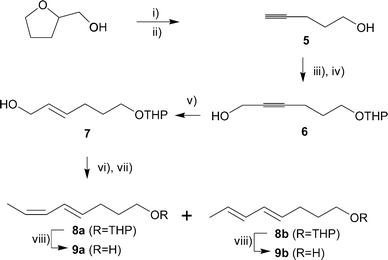 | ||
Scheme 1
Reagents and conditions: i) SOCl2, pyridine, 84%; ii) NaNH2, NH3 liq, 91%; iii) DHP, TsOH, CH2Cl2, 88%; iv) a) nBuLi, THF, −78 °C; b) (CHO)n, 84%; v) LiAlH4, Et2O, reflux, 85%; vi) a) DMSO, (COCl)2, DCM, −78 °C, then Et3N; b) Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH3, Et2O, −78 °C; vii) silica gel–AgNO3 chromatography, 22% 8a, 18% 8b; viii) TsOH, MeOH, 91% 9a, 89% 9b. CHCH3, Et2O, −78 °C; vii) silica gel–AgNO3 chromatography, 22% 8a, 18% 8b; viii) TsOH, MeOH, 91% 9a, 89% 9b. | ||
Alcohol 9a, obtained from subsequent THP deprotection of 8a, was converted into tetraene 3 in four steps as illustrated in Scheme 2, with the final Wittig reaction employing phosphonium salt 12,16 completing the installation of the isobutyl E,E-dieneamide moiety. This provided 3 as a white crystalline solid (mp 78–79 °C), previously only reported as an oil.17 The same methodology was also used to obtain tetraene 4 (mp 104–106 °C) from 9b, and constituted the first reported complete synthesis of this tetraene isomer. Tetraene 4 has been previously prepared by isomerisation (I2/UV) of a mixture of 1 and 2, that were isolated from Asiasarum heterotropoides,18 and the reported 13C NMR data agrees with our synthetic compound.
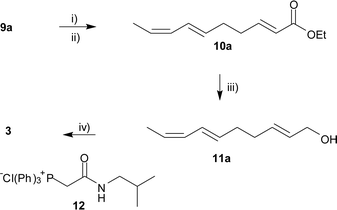 | ||
| Scheme 2 Reagents and conditions: i) DMSO, (COCl)2, CH2Cl2, −78 °C, then Et3N; ii) (EtO)2POCH2COOEt, NaH, Et2O, 71%; iii) DIBAL, Et2O, 87%; iv) a) DMSO, (COCl)2, CH2Cl2, −78 °C, then Et3N; b) 12, KOt-Bu, THF, 0 °C, 63%. | ||
When comparing our NMR data for 3 with the reported data for the naturally occurring compound isolated from S. mauritiana,6 we noted obvious discrepancies in both the 13C and 1H spectra. In particular the 13C chemical shift for C7 was reported as δ 26.97 ppm compared with δ 32.1 ppm in our synthetic standard. Upon comparison of the NMR data for all four synthetic tetraene isomers, it was found that the data for natural material isolated from S. mauritiana was in fact consistent with that of the Z,Z isomer 1, and not the E,Z isomer 3 as the authors had claimed.6 Inspection of the 13C NMR data from our synthetic tetraene standards revealed that the stereochemistry of the 8,10-diene moiety can be readily assessed by observation of the chemical shifts of C7 and C12, as shown in Table 1. The shielding experienced by allylic carbons adjacent to the cis double bond caused them to resonate approximately 5 ppm upfield compared with their trans counterparts, which is in accordance with literature observations.18
| Position | Tetraene 1 (ppm) | Tetraene 2 (ppm) | Tetraene 3 (ppm) | Tetraene 4 (ppm) |
|---|---|---|---|---|
| C7 | 26.7 | 26.9 | 32.1 | 31.8 |
| C12 | 13.1 | 18.3 | 13.3 | 18.0 |
One other reported isolation of tetraene 3 from Leucanthemum sp. has appeared in the literature,17 and although the 1H NMR data reported matched our data for 3, no 13C NMR or other physical data was available for the natural compound. Thus this report17 should be regarded as tentative and the 13C NMR data for 3 is reported correctly for the first time in this communication.
A synthesis of 3 has also previously been reported,19 but it contains no mention of stereochemical purity of the material obtained nor was 13C data supplied. Additionally, the 1H NMR data was stated to correlate with that of the incorrectly assigned material isolated from S. mauritiana. For this reason, and to supply a larger quantity of material for biological testing (vide infra), we undertook a stereospecific synthesis of 3. We proposed that a Suzuki coupling20 would provide the required stereocontrol in the preparation of the key alcohol intermediate 9a. Thus, the boronate ester 14 was prepared in excellent yield from protected pentynol 13. Upon coupling of 14 with cis-1-bromo propene under Suzuki conditions20 (Scheme 3), the diene 15 was produced as the sole isolated product and was simply converted to the desired alcohol (9a). This was readily converted to 3via the method already established (Scheme 2). In our hands, we found yields of the cross coupled product 15 to be very disappointing in the presence of a THP protecting group, but this was rectified by substitution with the TBS moiety.
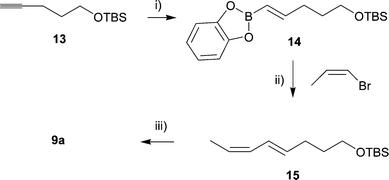 | ||
| Scheme 3 Reagents and conditions: i) catechol borane, 60 °C, 4 hr, 92%; ii) Pd(PPh3)4, NaOEt, benzene, 60 °C, 2 h, 77%; iii) MeOH, conc. HCl, 25 °C, 91%. | ||
With standards of the various alkyl amides 1–4 in hand, GCMS analysis of Echinacea extracts, along with co-injections with either 3 or 4, were undertaken. These studies revealed that the third tetraene present in these extracts (Fig. 1) was indeed the E,Z isomer 3 and that none of the E,E isomer 4 could be detected in any Echinacea sample examined. This prompted us to determine the relative proportions of the various tetraenes 1–4 in several E. angustifolia and E. purpurea root samples from Australia, Canada, Germany, New Zealand, USA and Kenya. Not surprisingly, the levels of tetraenes differed between the two species (Table 2). Unexpectedly, however, the relative abundance of 1–3 differed within a species, especially in E. purpurea (Table 2). However, not only did the levels of tetraenes in E. purpurea extracts differ greatly between samples grown in different locations but differences were found between plants grown in the same field! There did not appear to be any obvious geographical trends that could be correlated with the differences in respective tetraene levels.
| % in E. angustifoliaa (n = 6) | % in E. purpureaa (n = 10) | K i CB2/nM (n = 3) | |
|---|---|---|---|
| a Expressed as a percentage of the total tetraene alkyl amide content. In E. angustifolia this was 18.8 ± 9.8 mg g−1 and in E. purpurea this was 4.4 ± 4.0 mg g−1. | |||
| 1 | 10 ± 3 | 32 ± 14 | 57 ± 9 |
| 2 | 80 ± 5 | 62 ± 12 | 9044 ± 2985 |
| 3 | 10 ± 2 | 6 ± 6 | 4535 ± 711 |
| 4 | nd | nd | >100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
Due to the different abundance of the three isomers and the variation between the two Echinacea species, we wished to investigate the effect, if any, that double bond geometry had upon biological activity of these isomers 1–4. We chose to examine the binding affinity of each isomer for the CB2-receptor, as a recent study has shown that the tetraene alkyl amide 1 binds more strongly to this receptor than do endogenous cannabinoids.12
These studies revealed that tetraene 1 is a high affinity binder for the CB2 receptor as it competitively inhibited binding of the synthetic high affinity cannabinoid ligand [3H]CP55,940 (Table 1). However, tetraenes 2 and 3, whilst binding to CB2 exhibited much lower affinities, possessing Ki values approximately two orders of magnitude higher than isomer 1. Interestingly, tetraene 4, which has not been reported to occur naturally, showed no binding affinity for the CB2 receptor.
Conclusions
Concomitant use of GC/MS and pure synthetic tetraene alkyl amides, has allowed the positive identification of the (2E,4E,8E,10Z) tetraene isomer in Echinacea for the first time. The variation in the binding affinity for the CB2 receptor observed for the tetraene alkyl amides 1–3 raises the possibility that some of the differences seen in the activity of Echinacea preparations may be linked to the species used (E. purpurea or E. angustifolia) and the fluctuating ratios of tetraenes that would be encountered. Reporting the total tetraene content of an Echinacea preparation may not be specific enough, and quantification of the individual isomers may be required to truly allow comparison of biological data with different extracts. It is expected that the short stereoselective synthesis of isomerically pure 3 and 4 reported here will aid in providing pure material as a reference for the standardization of Echinacea products.Experimental
General conditions
All reactions involving air or moisture sensitive reagents were carried out under a nitrogen atmosphere in oven pre-dried glassware. Diethyl ether and THF were distilled from sodium–benzophenone prior to use; and dichloromethane was distilled from calcium hydride. CDCl3 and C6D6 were purchased from Cambridge Isotope Laboratories. NMR was performed on either a Bruker Avance 300 MHz, AV400 MHz or DRX500 MHz. Chemical shifts (δ) were referenced to internal solvent and are reported relative to SiMe4. GC/MS analyses were performed using a Shimadzu-17A GC equipped with J & W Scientific DB5 column (internal diameter 0.2 mm/30 m) coupled to a Shimadzu QP5050 Mass Spectrometer (70 eV). Echinacea extracts obtained from Mediherb Pty. Ltd. were prepared for GC/MS analysis by the following general proceedure: 0.5 ml of solution was stripped of solvent under vacuum (<0.1 mm) without external heating. Once the solvent was removed (1–2 mins) the residue was re-dissolved in methanol (0.1 ml) and refrigerated until analysis. GC parameters were as follows: t = 0–2 mins oven temp = 100 °C; t = 2–11.375 mins oven temp = 100–250 °C, ramped at 16 °C/min; t = 11.375–21 mins oven temp = 250 °C; t = 21 mins end run. Injector temp = 250 °C; interface temp = 270 °C; flow rate = 76.9 ml min−1 helium; split ratio = 22; injection volume = 2 µl.Butyllithium (2.46 ml, 3.2 mmol of a 1.3M solution in hexanes) was added dropwise to a suspension of ethyltriphenylphosphonium iodide (1.50 g, 3.6 mmol) in ether (20 ml) at room temperature. The resulting deep orange solution was stirred for a further 25 minutes, then cooled to 0 °C using an external ice-bath. Next the crude aldehyde prepared above was dissolved in ether (5 ml) and added dropwise over 2 minutes. After stirring a further 20 minutes at 0 °C, water (20 ml) was added and the mixture stirred vigorously for 5 minutes. The water was separated and the ether layer washed with brine, then dried (MgSO4) and evaporated. Then the residue was taken up in ether–hexane (1 : 1, 50 ml) and passed through a short column of silica gel in order to remove Ph3PO. The silica was rinsed well with solvent and the combined filtrates were evaporated to leave a colourless oil containing a roughly 1 : 1 mixture of 8a and 8b (220 mg) as evidenced by GC/MS. Separation of the isomers was achieved by column chromatography using silver nitrate impregnated silica gel prepared from AgNO3 (2.9 g) and silica gel 60 (22 g). Elution with 5% ethyl acetate in hexanes afforded 8b (92 mg, 18%) which eluted first, followed by 8a (113 mg, 22%).
8b δ H(300 MHz; CDCl3; Me4Si): 1.43–1.90 (m, 11H), 2.14 (q, 2H, J = 7.8 Hz), 3.33–3.53 (m, 2H), 3.69–3.90 (m, 2H), 4.58 (t, 1H, J = 3.0 Hz), 5.50–5.62 (m, 2H), 5.95–6.07 (m, 2H). δC(75 MHz; CDCl3; Me4Si): 17.9, 19.6, 25.6, 29.2, 29.5, 30.7, 62.2, 66.9, 98.8, 126.8, 130.7, 131.1, 131.6. δH(500 MHz; C6D6; Me4Si): 1.20–1.41 (m, 3H), 1.51–1.81 (m, 5H), 1.58 (d, 3H, d, J = 7.0 Hz), 2.14 (q, 2H, J = 7.5 Hz), 3.31 (dt, 1H, J = 6.5, 9.5 Hz), 3.35–3.41 (m, 1H), 3.76–3.82 (m, 1H), 3.79 (dt, 1H, J = 6.5, 9.5 Hz), 4.56 (dd, 1H, J = 3.0, 3.0 Hz), 5.99–6.08 (m, 2H), 5.47 (dq, 1H, J = 7.0, 14.0 Hz), 5.51 (dt, 1H, J = 7.0, 14.0 Hz). HRMS EI(m/z): [M+] calcd. for C13H22O2, 210.1619; found, 210.1622.
8a δ H(300 MHz; CDCl3; Me4Si): 1.42–1.87 (m, 11H), 2.20 (q, 2H, J = 7.2 Hz), 3.32–3.52 (m, 2H), 3.70–3.39 (m, 2H), 4.59 (t, 1H, J = 3 Hz), 5.38 (dq, 1H, J = 7.2, 11.1 Hz), 5.68 (dt, 1H, J = 7.5, 15.0 Hz), 5.98 (tq, 1H, J = 1.2, 11.1 Hz), 6.36 (ddq, 1H, J = 1.2, 10.8, 15 Hz). δC(75 MHz; CDCl3; Me4Si): 13.2, 19.6, 25.5, 29.4, 29.5, 30.7, 62.2, 66.9, 98.8, 124.0, 125.8, 129.4, 133.5. HRMS EI(m/z): [M+] calcd. for C13H22O2, 210.1619; found, 210.1618.
NaH (34 mg, 50% suspension in mineral oil, 0.7 mmol) was suspended in ether (5 ml) and cooled to −10 °C. Then (diethoxy-phosphoryl)-acetic acid ethyl ester (157 mg, 0.7 mmol) was added dropwise (H2 evolution) with stirring. After complete addition the mixture was stirred a further 10 minutes and the crude aldehyde solution prepared above was added slowly. After stirring a further 10 minutes at −10 °C, water (10 ml) was added. The organic layer was collected and the aqueous phase was extracted with hexanes. The combined organic layers were washed thoroughly with water and then once with brine. Drying (MgSO4) and evaporation of the solvents left an oil which was purified by column chromatography (silica gel 60, 10% ether in pentane) to give the title compound (48 mg, 77% over 2 steps) as a colourless oil. δH(400 MHz; CDCl3; Me4Si): 1.23 (t, 3H, J = 7.2 Hz), 1.68 (d, 3H, J = 6.4 Hz), 2.13–2.28 (m, 4H), 4.13 (q, 2H, J = 7.2 Hz), 5.41–5.60 (m, 2H), 5.78 (dt, 1H, J = 1.6, 15.2 Hz), 5.91–6.02 (m, 2H), 6.90 (dt, 1H, J = 7.2, 15.2 Hz). δC(100 MHz; CDCl3; Me4Si): 14.1, 17.8, 30.8, 31.9, 59.9, 121.5, 127.5, 129.5, 131.2, 131.3, 148.1, 166.4. HRMS EI(m/z): [M+] calcd. for C12H18O2, 194.1307; found, 194.1307.
KOt-Bu (39 mg, 0.35 mmol) was added in one portion to a stirred suspension of 1216 (165 mg, 0.4 mmol) in THF (3 ml) at room temperature. Stirring was continued for a further 20 minutes at this temperature, then the mixture was cooled to 0 °C and the crude aldehyde solution was added in one portion. Stirring was continued at 0 °C for 30 minutes, then saturated ammonium chloride solution (7 ml) was then added and the organic phase was separated. The aqueous phase was extracted into ether, and the combine organic phases washed with brine, dried (MgSO4) and concentrated in vacuo. Purification by column chromatography (silica gel 60, 20% ethyl acetate in hexane) yielded the title compound (19 mg, 40%) as a white solid. Mp = 104–106 °C. δH(500 MHz; CDCl3; Me4Si): 0.92 (d, 6H, J = 6.8 Hz), 1.72 (d, 3H, J = 6.4 Hz), 1.79 (sept, 1H, J = 6.8 Hz), 2.15–2.27 (m, 4H), 3.16 (dd, 2H, J = 6.4, 6.4 Hz), 5.46–5.64 (m, 3H), 5.76 (d, 1H, J = 15.2 Hz), 5.96–6.09 (m, 3H), 6.14 (dd, 1H, J = 10.4, 15.2 Hz), 7.17 (dd, 1H, J = 10.4, 15.2 Hz). δC(125 MHz; CDCl3; Me4Si): 18.0, 20.1, 28.6, 31.8, 32.8, 46.9, 122.1, 127.5, 128.6, 130.3, 131.1, 131.4, 141.1, 142.0, 166.3. Anal. Calcd: C, 77.68; H; 10.19; N, 5.66; Found C 77.46; H, 10.18; N, 5.84%.
References
- R. Bauer, P. Remiger and H. Wagner, Phytochemistry, 1988, 7, 2339–2342 Search PubMed; R. Bauer, P. Reminger and H. Wagner, Phytochemistry, 1989, 28(2), 505–508 CrossRef CAS.
- W. Herz and P. Kulanthaivel, Phytochemistry, 1985, 24(1), 173–174 CrossRef CAS.
- F. Zhang, C.-H. Chu, Q. Xu, S.-P. Fu, J.-H. Hu, H.-B. Xiao and X.-M. Liang, J. Asian Nat. Prod. Res., 2005, 7(1), 1–5 Search PubMed.
- H. Garcia-Barriga, Flora Medicinal de Colombia, Botánica Médica, Vol. 3, Universidad Nacional de Columbia, Bogotá, Columbia, 1975 Search PubMed.
- F. Zhang, Q. Xu, S. Fu, X. Ma, H. Xiao and X. Liang, Flavour Fragrance J., 2005, 20, 319–320.
- I. J. O. Jondiko, Phytochemistry, 1986, 25(10), 2289–2290 CrossRef CAS.
- M. Hall and N. Nazir, Alternative Therapies, 2005, 11(3), 34–37 Search PubMed.
- B. Barret, Phytomedicine, 2003, 10, 66–86 Search PubMed.
- R. B. Turner, R. Bauer, K. Woelkart, T. C. Hulsey and J. D. Gangemi, N. Engl. J. Med., 2005, 353(4), 341–348 CrossRef CAS.
- A. Matthias, J. T. Blanchfield, K. G. Penman, I. Toth, C. S. Lang, J. J. DeVoss and R. P. Lehman, J. Clin. Pharm. Ther., 2004, 29, 7–13 CrossRef CAS.
- S. Merali, S. E. Binns, P. Levasseur, C. Ficker, M. L. Smith, B. R. Baum, E. Brovelli and J. T. Arnason, Pharm. Biol. (Lisse, Neth.), 2003, 41, 412–420 CrossRef CAS; L. J. Clifford, M. G. Nair, J. Rana and D. L. Dewitt, Phytomedicine, 2002, 9, 249–253 Search PubMed.
- S. Raduner, A. Majewska, J.-Z. Chen, X.-Q. Xie, J. Hamon, B. Faller, K.-H. Altmann and J. Gertsch, J. Biol. Chem., 2006, 281(20), 14192–14206 CrossRef CAS.
- M. Hudaib, J. Fiori, M. G. Bellardi, C. Rubies-Autonell and V. Cavrini, J. Pharm. Biomed. Anal., 2002, 29, 1053–1060 CrossRef CAS.
- L. Brandsma, Preparative Acetylenic Chemistry, Elsevier, Amsterdam, 2nd edn, 1988, pp. 182–183 Search PubMed.
- T. S. Li, J. T. Li and H. Z. Li, J. Chromatogr., 1995, 715, 372–375 CrossRef CAS.
- M. Elliott, A. W. Farnham, N. F. Janes, D. M. Johnson and D. A. Polman, Pestic. Sci., 1987, 18(3), 191–201 CrossRef CAS.
- F. Bohlmann, U. Fritz and L. Dutta, Phytochemistry, 1980, 19, 841–844 CrossRef CAS.
- I. Yasuda, K. Takeya and H. Itokawa, Chem. Pharm. Bull., 1981, 29(2), 564–566 CAS.
- G. V. M. Sharma, T. Shekharam and V. Upender, Tetrahedron, 1990, 46(16), 5665–5672 CrossRef CAS.
- N. Miyaura, K. Yamada, H. Suginome and A. Suzuki, J. Am. Chem. Soc., 1985, 107, 972–980 CrossRef CAS.
- J. A. Marshall, J. Grote and J. E. Audia, J. Am. Chem. Soc., 1987, 109, 1186–1194 CrossRef CAS.
- J. A. Marshall and B. S. Deltoff, J. Org. Chem., 1986, 51(6), 863–872 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |