Exploring the chemistry of penicillin as a β-lactamase-dependent prodrug†
Carol C.
Ruddle
and
Timothy P.
Smyth
*
Department of Chemical and Environmental Sciences, University of Limerick, National Technological Park, County Limerick, Ireland. E-mail: timothy.smyth@ul.ie
First published on 22nd November 2006
Abstract
The penam nucleus can be modified to behave as a β-lactamase-dependent ‘prodrug’ by incorporation of a vinyl ester side chain at the 6-position. Enzyme-catalysed hydrolysis of the β-lactam ring uncovers the thiazolidine-ring nitrogen as a nucleophile that drives a rapid intramolecular displacement on the side chain. Attachment of 7-hydroxy-4-methylcoumarin as the releasable group of this side chain generated a penicillin structure that can function as a fluorescence-based reporter substance/diagnostic for the presence of low levels of β-lactamase enzyme in solution. Mechanistic details of the reaction pattern are documented and the scope and limitations of exploiting the structural modification are discussed.
Introduction
β-Lactams remain the frontline antibiotics for combating infections caused by Gram-negative bacteria. Their efficacy, however, is being compromised increasingly by the dissemination of β-lactamase enzymes throughout this bacterial population.1 While many serine-based β-lactamases are susceptible to inhibition by compounds such as clavulanic acid and sulbactam, the emergence of metallo-β-lactamases poses a new challenge, as these are not susceptible to such inhibitors, and the newer generation of these enzymes act on a broad variety of β-lactam structures—penicillins, cephalosporins and carbapenems. β-Lactamase-dependent prodrugs provide a potential strategy to counter the spread of β-lactamases by exploiting their presence, rather than blocking their action.2 Both the cephalosporin and carbapenem nucleus incorporate an inherent reactivity pattern that allows these to be readily configured as β-lactamase-dependent prodrugs. Thus, elimination of the 3′-substituent in cephalosporins (Scheme 1a), and of the 2′-substituent in carbapenems, is triggered by scission of the β-lactam; this reactivity has been explored mainly in the development of cephalosporin–quinolone conjugates.3,4 Despite the accumulation of detailed biological activity data, the cephalosporin–quinolone conjugates have not been launched as therapeutic agents.3f Nonetheless, the strategy of developing β-lactamase-dependent prodrugs remains valid and, in this context, we have explored the chemistry of the penam nucleus with the aim of incorporating a latent ‘prodrug’ reactivity that is triggered by scission of the β-lactam ring. A generic modification that achieves this is attachment of an unsaturated linker, with an electron-deficient centre bearing a moderately nucleofugic group, at the 6-position of penicillin. Structures 1 and 2 are prototypic examples: enzyme-catalysed scission of the β-lactam ring brings two potential nucleophiles into play—the thiazolidine-ring sulfur and nitrogen atoms—resulting in a rapid intramolecular reaction with loss of the nucleofugic group (Scheme 1b).5,6 The efficacy of the displacement is due to the syn juxtaposition of the nucleophile and electron-deficient centre on the unsaturated linker6 of the ring-opened structure, leading to formation of a five-membered ring: this type of molecular construct gives rise to reactions of very high effective molarity.7 It proved difficult to prepare S-aminosulfenimino structures (Scheme 1, 1) with leaving groups other than a small set of arylsulfonamides.8 As a result we turned our attention to penicillin structures bearing a vinylester side chain. Penicillin structures bearing this side chain, and with the thiazolidine ring sulfur in the sulfone oxidation state, have previously been found to act as β-lactamase inhibitors, whereas the corresponding sulfides are not inhibitors.9a In the context of our work, the vinyl ester side chain was seen as an alternative to the S-aminosulfenimine moiety and one offering better scope for synthetic variation of the leaving group. In a preliminary communication6 we reported on the preparation and reaction of a variety of compounds of structure type 2 bearing distinct leaving groups; in this paper we expand on the chemistry of these penicillins and on the scope and limitations of their capability to act as β-lactamase-dependent prodrugs.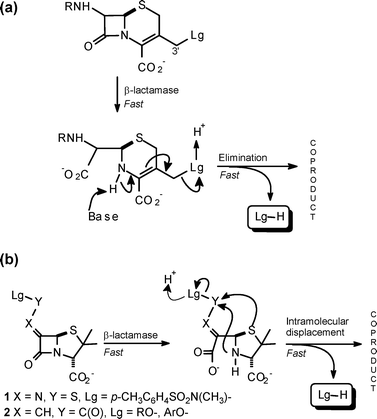 | ||
| Scheme 1 β-Lactams showing a latent prodrug reactivity pattern: (a) a typical cephalosporin, (b) generic penicillin structures. | ||
Results and discussion
An efficient route to structure type 2 is via reaction of oxo-penicillin derivative 3 with a Wittig reagent (Scheme 2) as originally developed by Buynak.9 The benzhydryl esters 4–7 were prepared using commercially available Wittig reagents and, where required, the corresponding sodium salts were obtained by removal of the benzhydryl group.6 Preparation of 8′ was achieved by removal of the allyl group of 5 followed by DCC mediated coupling of the resulting free acid with 7-hydroxy-4-methylcoumarin (below referred to as coumarin) and, finally, removal of the benzhydryl group. This structure is a potential reporter molecule/diagnostic for the presence of β-lactamase enzyme as 8′ is non-fluorescent, whereas coumarin itself is highly fluorescent.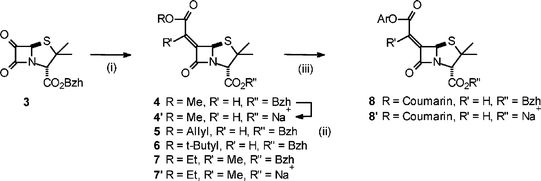 | ||
Scheme 2 Synthetic route: (i) ROC(O)CR′![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3, −55 °C; (ii) (a) AlCl3, −84 °C, (b) NaHCO3, freeze dry; (iii) (a) 5, Pd(PPh3)4, sodium p-toluene sulfinate, THF–H2O, (b) 7-hydroxy-4-methylcoumarin, DCC. PPh3, −55 °C; (ii) (a) AlCl3, −84 °C, (b) NaHCO3, freeze dry; (iii) (a) 5, Pd(PPh3)4, sodium p-toluene sulfinate, THF–H2O, (b) 7-hydroxy-4-methylcoumarin, DCC. | ||
The reaction pattern shown in Scheme 3a was initially validated for the benzhydryl esters 4 and 5 in a reaction system of CH3OH–0.2 M NEt3 for 40 min at r.t. (nominally 18 °C); the ring-fused γ-lactam 10 was formed exclusively and was fully characterised.6 This isomer, with the unsaturated bridgehead position, was found to be 3.74 kcal mol−1 more stable (optimised structures, 6–311G level, Gaussian 03) than 9; the isomerisation is driven, presumably, by initial deprotonation at the bridgehead carbon of 9 in the basic reaction medium. With the t-butyl derivative 6 the reaction stopped after ring opening (6a was isolated and characterised);6 it would appear that the size of t-butyl group impedes the intramolecular displacement. The β-lactam ring of the ethylidene structure 7 was remarkably stable and remained totally unchanged under the above reaction conditions; ester exchange of the benzhydryl group was the only process observed after 3 h. This stability was also observed for the corresponding sodium salt 7′ in aqueous buffer (see below) and indicates that the exocyclic methyl group, juxtaposed syn to the lactam carbonyl, provides a significant barrier to nucleophilic attack. Indeed, the tetrahedral intermediate formed by addition of methoxide to the α-face of an ethylidene β-lactam unit was found to be 2.1 kcal mol−1 less stable (optimised structures, 6–311G level, Gaussian 03) than the corresponding methylidene intermediate, when compared with the respective starting materials.10 Such a difference in activation energies would lead to a reduction in reaction rate of over two orders of magnitude. (The ethylidene structural feature is present in asparenomycins11—naturally occurring carbapenem structures—and may contribute to their stability.)
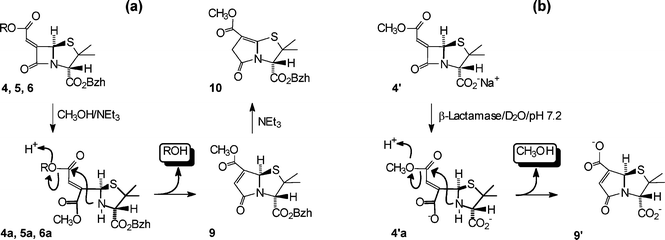 | ||
| Scheme 3 Reaction pattern of: (a) benzhydryl esters in CH3OH–0.2 M NEt3, (b) sodium salt 4′ in D2O buffer (pH 7.2), 25 °C in the presence of β-lactamase enzyme. | ||
The reactivity patterns of the salts 4′, 7′ and 8′ in aqueous buffer (D2O, pH 7.2, 25 °C) both in the absence and presence of β-lactamase enzyme were examined and the results are presented below. In the absence of enzyme 4′ had a half-life of 5 days. The reaction products were identical to those of the enzyme-catalysed hydrolysis (see following), which confirms that the β-lactam ring is hydrolysed more readily that the aliphatic ester group. The time course of the reaction of 4′ in the presence of β-lactamase enzyme was followed by 1H NMR spectroscopy (see Fig. 1). The spectral sequence shows the initial formation of an intermediate and conversion of this to a single final product with the co-production of methanol. The key indicator that the intermediate is the ring-opened structure 4′a, is the large upfield shift in the α-CH3 resonance6 (Fig. 1b,c); there is also a small upfield shift in the methyl ester resonance and a distinct change in the H-5 and H-8 resonances. The conversion of 4′a to 9′ with the concomitant product of methanol is clearly seen in the sequence of spectra (b) → (c) → (d) (Fig. 1). For the end product (Fig. 1d), the downfield shift of the α-methyl resonance (compared with that of the ring-opened structure 4′a) is notable and is consistent with the re-formation of a ring-fused bicyclic structure, while the relatively short reaction time for the whole transformation precludes simple hydrolysis of the methyl ester group of 4′a. The 13C-NMR and high-resolution mass spectra of the product isolated after workup are consistent with structure 9′.
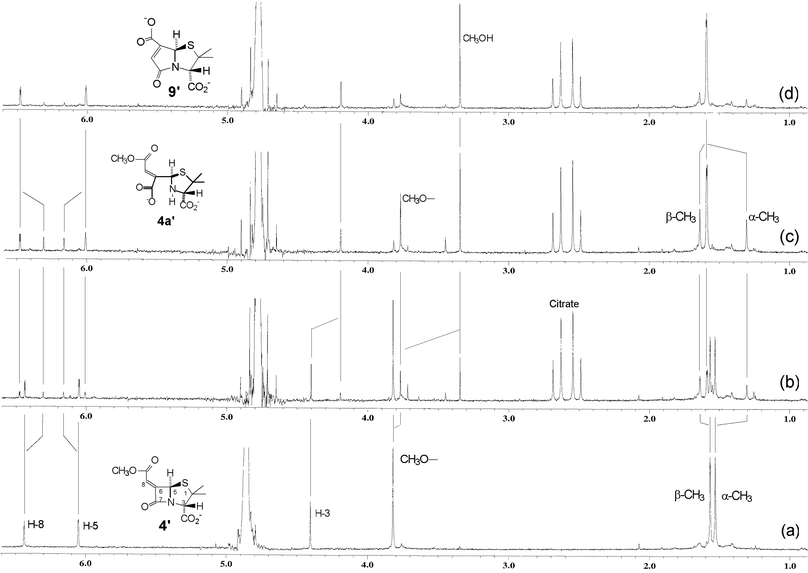 | ||
| Fig. 1 1H NMR spectra of (a) 4′ in D2O buffer (0.02 M phosphate, pH 7.2, 25 °C) and (b) 7.5 min, (c) 38 min, (d) 50 min following addition of β-lactamase enzyme in a citrate buffer. | ||
Unlike the case of the benzhydryl esters 4 and 5, the γ-lactam isomer obtained here was 9′; the aqueous reaction medium was not sufficiently basic to deprotonate the bridgehead carbon atom and, in addition, the carboxylate side chain should impede the deprotonation.
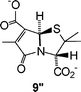
The ethylidene salt 7′ showed remarkable stability in aqueous buffer and was unchanged after 14 days (this parallels the earlier observation on the stability of β-lactam ring of ester 7 in CH3OH–0.2M NEt3). Nonetheless, β-lactamase-catalysed hydrolysis of 7′ was effective and this led cleanly to the formation of 9″ and the co-production of ethanol. (The formation of 9″ confirms the Z stereochemistry of the side chain of 7′.) During the course of the reaction build up, the corresponding ring-opened structure was not observed.10 It is interesting to note that the vinyl methyl group, which exerts such a dramatic effect on nucleophilic attack at the lactam carbonyl, does not impede the intramolecular nucleophilic displacement at the side chain carbonyl.
β-Lactamase-catalysed hydrolysis of 8′ led cleanly to the formation of 9′ and the co-release of coumarin; the corresponding ring-opened structure was not observed during the course of the reaction. In the absence of enzyme 8′ was much more labile than the aliphatic derivative 4′, giving a half-life of 90 min in the buffer system. This lability was associated with direct hydrolysis of the aromatic ester side chain rather than with hydrolysis of the β-lactam ring. The 1H NMR spectra of the end products of hydrolysis of 8′ in the absence and presence of β-lactamase enzyme are shown in Fig. 2: in the presence of enzyme the products were coumarin and γ-lactam 9′, while in the absence of enzyme the products were coumarin and β-lactam 11 (the high-resolution mass spectra of the product isolated after workup is consistent with structure 11 (see the Experimental section)). A summary of some of the key data on 4′, 7′ and 8′ is given in Table 1.
| Structure | Relative rate of turnover by β-lactamase enzymea | Half-life in 0.02 M phosphate buffer, pH 7.2, 25 °C |
|---|---|---|
| a Penicillinase Type 1 from B. cereus. b The specific activity with benzylpenicillin was 1872 µmol min−1 mg−1 protein. c Hydrolysis of the β-lactam ring. d No hydrolysis occurred within 14 days. e Hydrolysis of the aryl ester side chain. | ||
| 4′ | 1 | 7 daysc |
| 7′ | 0.5 | ≫14 daysd |
| 8′ | 20 | 1.5 he |
| Benzylpenicillin | 1260b | — |
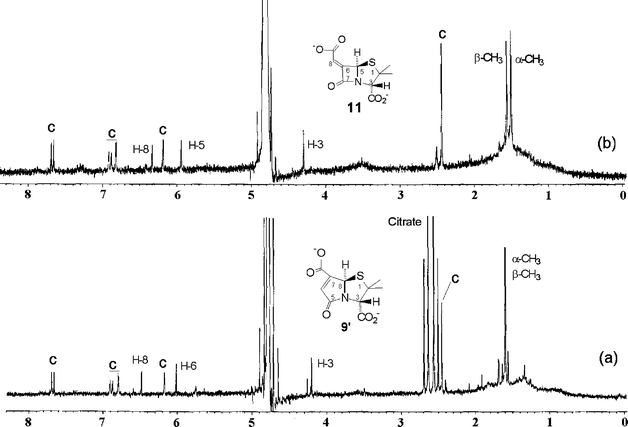 | ||
| Fig. 2 1H NMR spectra of the hydrolysis products of 8′ (a) 50 min after addition of β-lactamase enzyme, and (b) after 12 h in D2O buffer (0.02 M phosphate, pH 7.2, 25 °C). The peaks labelled C correspond to free 7-hydroxy-4-methylcoumarin in both spectra. | ||
The release of coumarin from 8′ was also monitored by fluorescence spectroscopy. The time course of the fluorescence emission of a 1.4 mM solution of 8′ (phosphate buffer, pH 7.2), when treated with various amounts of β-lactamase enzyme, is shown in Fig. 3. It is clear that 8′ can act as a reporter molecule for the presence of quite low amounts (∼6 units)12 of β-lactamase enzyme in solution. When 8′ was treated with a solution containing β-lactamase-producing E. coli cells13 the rate of fluorescence emission was not faster than that associated with background hydrolysis of 8′ itself, whereas, when treated with cell lysate the rate of fluorescence emission was considerably faster. These observations indicate that 8′ cannot efficiently permeate the outer membrane of E. coli.
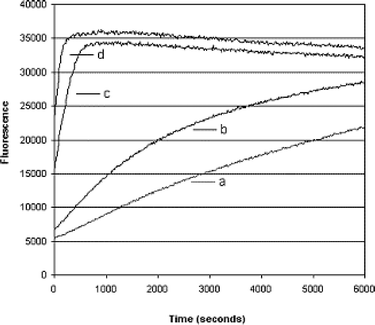 | ||
| Fig. 3 Increase in fluorescence emission as a function of time observed for a 1.4 mM solution of 8′ in the presence of (a) 0, (b) 6, (c) 30, and (d) 60 units of β-lactamase enzyme (penicillinase type 1). | ||
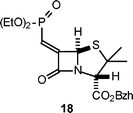
Although aryl esters are more readily hydrolysed than aliphatic esters, the lability of the ester side chain of 8′, compared to that of 4′, was unexpected. The relatively short half-life of 8′ in aqueous buffer was a drawback as it restricted the type of releasable prodrug group that could be usefully incorporated and evaluated (e.g. triclosan). It was envisaged that an aryl sulfonate side chain would avoid this limitation and that such a structure might be obtainable via a Horner–Wittig–Emmons reaction of the required sulfophosphonate with 3 (Scheme 4). Whereas this method was successful in producing the alkyl sulfonate derivative 16 (26% yield) it did not yield the aryl sulfonate 17. The problem was the lack of reactivity of phenylmethanesulfonate anion as a nucleophile in general, and specifically toward diphenylchlorophosphate14 to produce 15. Treatment of 13 with n-BuLi followed by CH3I failed to produce any methylated product (a similar process with 12 rapidly produced methylethane sulfonate). The breakdown of 13 in the presence of n-BuLi to generate sulfene15 and phenoxide was not the reason for this lack of nucleophilic behaviour as treatment of 13 with n-BuLi followed by quenching with D2O lead to the recovery of a substantial amount of deuterated phenylmethane sulfonate. As a similar problem is likely to be associated with the anion of 15, this approach was not pursued further. A phosphonate side chain was considered as a final alternative. Treatment of 3 with the anion derived from tetraethylbisphosphonate produced the penicillin derivative 18; as this was obtained in only a 1% yield (after purification) no further evaluation was undertaken.
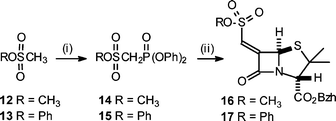 | ||
| Scheme 4 (i) (a) n-BuLi, (b) Cl(O)P(OPh)2, −78 °C; (ii) (a), n-BuLi, (b) 3, −78 °C. | ||
The release of methanol from the cephalosporin salts 19′ and 20′ was also studied and the key findings are summarised in Scheme 5. Cephalosporin 19′ was as stable as the corresponding penicillin 4′ with respect to background hydrolysis and also behaved as a β-lactamase substrate. Loss of methanol from the ring-cleaved structure 19′a was considerably faster than that observed in background hydrolysis, thus indicating anchimeric assistance. The finding that loss of methanol from the ring-cleaved sulfoxide 20′a was slower than from 19′a, identifies the dihydrothiazine sulfur as the nucleophile providing this assistance, and not the imine nitrogen; the coproduct(s) were not identified. Treatment of the desacetoxy cephalosporin salt 21′ with β-lactamase enzyme led quantitatively to the release of methanol and the formation of the γ-lactam 22 (Scheme 6); the ring-opened structure was observed here as it was with 4′a.10 With a half-life of 55 min for the displacement the enamine nitrogen of 21′a is more nucleophilic, toward the side chain carbonyl, than the dihydrothiazine sulfur, but is less nucleophilic by about a factor of 5, that the amine nitrogen in the ring-opened penicillin 4′a.
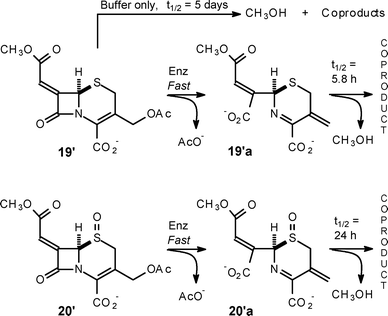 | ||
| Scheme 5 Rates of methanol release from cephalosporin structures 19′ and 20′. | ||
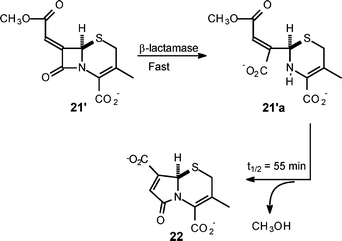 | ||
| Scheme 6 γ-Lactam formation from desacetoxy cephalosporin structure 21. | ||
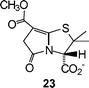
γ-Lactam salt 23, obtained from 10 by removal of the benzhydryl group, was screened for antibiotic and β-lactamase-inhibitory activity. While no antibiotic activity was observed, 23 was found to show weak inhibition (50% at 100 µM) of the metallo-β-lactamase CphA from Aeromonas hydrophilia.16
Conclusions
Engineering the penicillin nucleus to act as a viable β-lactamase-dependent prodrug presents a distinct challenge compared with that of achieving the same capacity in cephalosporins. Ideally, the intact structure must behave as an antibiotic in its own right and, in addition, the ‘prodrug’ component must behave as an independent antibiotic when released following scission of the β-lactam ring. The 7-amino- and 3′-substituents of cephalosporins makes it easier to achieve this: the 7-amino substituent (not-releasable) can be used to obtain normal β-lactam antibiotic activity, while the 3′-substituent can be chosen to act as an independent antibiotic when released. In the case of a penicillin bearing either a vinyl ester or S-aminosulfenimine side chain (Scheme 1; 1 and 2, respectively), this group must allow for normal β-lactam antibiotic activity of the intact structure, and it must also carry the releasable moiety that can function as an independent antibiotic. In all cases the releasable antibiotic component must have biological activity and clearance profiles that are compatible with those of a β-lactam; this is a concern in the case of the cephalosporin–quinolone conjugates.5c In our work we have identified the chemical basis of modifying the penam unit so as to incorporate a latent prodrug reactivity that is triggered by the action of a β-lactamase enzyme. One functional implementation of this is the coumarin derivative 8′ which can act as a fluoresence-based reporter molecule for the presence of low levels of β-lactamase enzyme in solution.Experimental
Benzhydryl-6-(Z)-[1-ethoxycarbonylethylidene]penicillanate (7)
Benzhydryl-6-oxopenicillinate 36 (1.04 g, 2.73 mmol) was dissolved in dichloromethane (13 mL). The solution was cooled to −55 °C under nitrogen. To this cooled solution (1-ethoxycarbonylethyledene)triphenylphosphorane (931.4 mg, 2.57 mmol) in dichloromethane (30 mL) was added dropwise over 30 min. Stirring was continued for a further 10 min. Analysis by TLC (50 : 50, ethyl acetate–hexane) indicated disappearance of the 3 and the formation of a new product of higher Rf value. The solution was allowed warm to room temperature and was washed with water (20 mL), the organic layer was separated, dried and concentrated under reduced pressure to leave crude 7. Purification by chromatography (silica gel, hexane–ethyl acetate 60 : 40) yielded 7 as a waxy, yellow oil (737 mg, 1.63 mmol, 63%). ESI-HRMS for C26H27NO5S: [M + H]+1 calcd: 466.1688, found: 466.1709. δH (270 MHz, CDCl3) 1.27 (s, 3H, α-CH3), 1.33 (t, J = 7.2 Hz, 3H, CH2CH3) 1.56 (s, 3H, β-CH3), 2.20 (s, 3H, CH3), 4.25 (q, J = 7.2 Hz, 2H, CH2CH3) 4.64 (s, 1H, H-3), 5.95 (s, 1H, H-5), 6.93 (s, 1H, CH(Ar)2), 7.22–7.42 (m, 10H, ArH); δC (67.93 MHz, CDCl3) 14.13, 14.45, 25.59 (CH3CH2O, CH3CH, α-CH3), 33.67 (β-CH3), 61.77, 63.41, 69.89, 70.34 (C-2, C-3, C-5, CH3CH2O), 78.31 (CH(Ar)2), 127.03, 127.52, 128.16, 128.34, 128.55, 128.62 (C, Ar), 139.17, 139.23 (C, Ar), 149.05 (C-6), 165.10, 167.02, 167.82 (3 × CO).
Sodium-6-(Z)-[1-ethoxycarbonylethylidene]penicillanate (7′)
7 (200 mg, 0.442 mmol) was dissolved in dichloromethane (16 mL) and was cooled under nitrogen to −84 °C. A solution of aluminium trichloride (240 mg, 1.80 mmol) in nitromethane (1.5 mL) was added in one portion to the cooled penicillin solution at which point the solution became intensely yellow. After stirring for 20 min, ethyl acetate (75 mL) and 5% sodium hydrogen carbonate (75 ml) were added successively whilst maintaining the temperature at −84 °C. The resulting slushy mixture was allowed to reach room temperature, the two layers were separated and the aqueous layer was filtered through celite. The filtrate was extracted with ethyl acetate (20 ml). The aqueous extract was collected and ethyl acetate (30 mL) was layered on top, the pH was adjusted to 2.2, and extracted with the ethylacetate. The aqueous layer was extracted with a second portion of ethyl acetate (30 mL). The organic extracts were combined, dried and concentrated under reduced pressure to leave the free acid of 7 as a pale yellow solid (98 mg, 0.305 mmol, 69%). ESI-HRMS for C13H17NO5S: [M–H]−1 calcd: 298.0749, found: 298.0744; δH (300 MHz, CDCl3) 1.35 (t, J = 6.9 Hz, 3H, CH2CH3) 1.58 (s, 3H, α-CH3), 1.62 (s, 3H, β-CH3), 2.21 (s, 3H, CH3), 4.27 (m, J = 6.9 Hz, 2H, CH2CH3) 4.57 (s, 1H, H-3), 5.93 (s, 1H, H-5), 8.11 (br s, 1H, OH). The free acid of 7 (83 mg, 0.258 mmol) was dissolved in ethyl acetate and extracted with aqueous sodium hydrogen carbonate (20.6 mg, 0.245 mmol) and the resulting aqueous layer was lyophilised to leave 7′ as a yellow solid (67.2 mg, 0.209 mmol, 85%) δH (270 MHz, D2O) 1.30 (t, J = 7.2 Hz, 3H, CH2CH3) 1.48 (s, 3H, α-CH3), 1.55 (s, 3H, β-CH3), 2.11 (s, 3H, CH3), 4.18–4.31 (m, 2H, CH2CH3) 4.33 (s, 1H, H-3), 5.96 (s, 1H, H-5).Sodium-6-(Z)-(methylumbelliferyl)carbonylmethylidenepenicillanate (8′)
Benzhydryl ester 86 (135 mg, 0.232 mmol) was treated as described above. The free acid of 8′ (73 mg, 0.175 mmol, 76%), recovered as a pale yellow solid, was dissolved in ethyl acetate and extracted with aqueous sodium hydrogen carbonate (13.25 mg, 0.157 mmol) and the resulting aqueous layer was lyophilised to leave 8′ as a yellow solid. δH (270 MHz, D2O) 1.54 (s, 3H, α-CH3), 1.61 (s, 3H, β-CH3), 2.51 (s, 3H, CH3Ar), 4.46 (s, 1H, H-3), 6.17 (s, 1H, H-5), 6.42 (s, 1H, C(O)CHC, side chain), 6.66 (s, 1H CHC, coumarin), 7.27 (dd, J = 1.86, 8.91 Hz, 1H, ArH, coumarin), 7.33 (d, J = 1.86 Hz, ArH, coumarin), 7.89 (d, J = 8.91 Hz, 1H, ArH, coumarin).
Diphenylphosphorylmethanesulfonate (14)
Methylmethane sulfonate (800 µL, 9.44 mmol) was dissolved in dry THF (24 mL) and the resultant solution was purged with nitrogen for 5 min. The solution was then cooled to −78 °C and 2 M n-BuLi in pentane (5.2 mL, 10.38 mmol) was added. Diphenylchlorophosphate (1076 µL, 5.2 mmol) was added after stirring the butyllithium solution at −78 °C for 15 min. The resultant solution was maintained at −78 °C for 30 min and then at −50 °C for 60 min. Ammonium chloride (555.44 mg, 10.58 mmol) in H2O (6 mL) was added and the mixture was warmed to room temperature. The mixture was diluted with water (50 mL) and extracted with dichloromethane (2 × 60 mL). The combined organic extracts were washed with water (3 × 40 mL), dried over MgSO4, filtered and concentrated to yield a white solid (1.82 g). The crude material was recrystallised from diethyl ether–dichloromethane to give the 14 as a white solid (1.04 g, 3.03 mmol, 58%). Mp 82–83.5 °C; found C, 48.87; H, 4.23%. C14H15O6PS requires C, 49.12; H, 4.42%; δH (270 MHz, CDCl3) 4.04 (s, 3H, CH3), 4.04 (d, J = 17.32 Hz, 2H, –CH2–P), 7.20–7.34 (m, 10H, ArH); δC (75.47 MHz, CDCl3) 46.94 (d, J = 144.28 Hz, CH2-P, inverted in DEPT135), 57.68 (CH3O), 120.61, 120.54, 126.03, 126.01, 130.06, 130.04, (C, Ar), 149.56, 149.69 (C, Ar).Benzhydryl-6-(Z)-methoxysulfonatemethylidenepenicillanate (16)
Diphenylphosphorylmethanesulfonate (14) (1.51 g, 2.57 mmol) was dissolved in THF (8 ml) and cooled under nitrogen atmosphere to −78 °C. 2 M n-BuLi (1.28 ml, 2.57 mmol) was added and the mixture was stirred for 15 minutes at −78 °C. 3 (1.04 g, 2.73 mmol) was added to the cold mixture and was allowed to stir at this temperature for a further 10 min. Analysis by TLC (50 : 50, ethyl acetate–hexane) indicated disappearance of the oxo-compound and the formation of a new product of higher Rf value. The solution was allowed warm to room temperature and was washed with water (20 mL), the organic layer was separated, dried and concentrated under reduced pressure to leave crude 16 as a brown oil. Purification by chromatography (silica gel, hexane–ethyl acetate, 70 : 30) yielded 16 (339.5 mg, 0.657 mmol, 26%). Mp 54–56 °C; found C, 58.42; H, 4.92; N, 2.94%. C23H23NO6S2 requires C, 58.34; H, 4.90; N, 2.96%; νmax (KBr)/cm−1 1779, 1745; δH (270 MHz, CDCl3) 1.26 (s, 3H, α-CH3), 1.57 (s, 3H, β-CH3), 3.92 (s, 3H, –OCH3), 4.47 (s, 1H, H-3), 6.04 (s, 1H, H-5), 6.68 (s, 1H, CH side chain), 6.95 (s, 1H, CH(Ar)2), 7.32–7.36 (m, 10H, ArH); δC (67.93 MHz, CDCl3) 25.26 (α-CH3), 33.73 (β-CH3), 57.47 (CH3O), 64.36, 68.46, 70.82 (C-2, C-3, C5), 78.68 (CH(Ar)2), 118.63 (side chain C(O)C), 127.04, 127.53, 128.31, 128.50, 128.63, 128.69 (C, Ar), 138.79, 138.89 (C, Ar), 156.08 (C-6), 163.98, 163.99, 166.19 (3 × CO).
Benzhydryl-6-(Z)-diethoxyphosphonatemethylidenepenicillanate (18)
Tetraethylmethylenediphosphonate (425.7 µL, 1.71 mmol) was dissolved in THF (8 mL) and cooled under nitrogen atmosphere to −78 °C. 2 M n-BuLi (0.856 mL, 1.71 mmol) was added and the mixture was stirred for 15 minutes at −78 °C. 3 (0.98 g, 2.56 mmol) was added to the cold mixture and was allowed to stir at this temperature for a further 10 minutes. Analysis by TLC (50 : 50, ethyl acetate–hexane) indicated disappearance of the oxo-compound. The solution was allowed warm to room temperature and was washed with water (20 mL), the organic layer was separated, dried and concentrated under reduced pressure to leave a brown oil. Purification by chromatography (silica gel, hexane–ethyl acetate, 70 : 30) gave a mixture of products. This purification step was repeated to yield a pure sample 18 (∼10 mg, ∼1%). ESI-HRMS for C26H30NO6PS: [M + Na]+1 calcd: 538.1429, found: 538.1422; δH (270 MHz, CDCl3) 1.26 (s, 3H, α-CH3), 1.35 (t, J = 5.44 Hz, 3H, –(OCH2CH3)A), 1.36 (t, J = 5.44 Hz, 3H, –(OCH2CH3)B), 1.56 (s, 3H, β-CH3), 4.08–4.21 (m, 4H –(OCH2CH3)A overlapping with –(OCH2CH3)B), 4.64 (s, 1H, H-3), 5.98 (t, J = 1.24 Hz, 1H, H-5), 6.24 (dd, J = 1.24, 13.36 Hz, 1H, CH sidechain), 6.95 (s, 1H, CH(Ar)2), 7.23–7.44 (m, 10H, ArH).Benzhydryl-7-[(Z)-(methoxycarbonyl)methylidene)]cephalosporanate-1-sulfoxide (20)
Benzhydryl-7-oxocephalosporanate, prepared as described for 3,6 (1.027 g, 2.35 mmol) was dissolved in dichloromethane (15 mL). The solution was cooled to −84 °C under nitrogen. To this cooled solution methyl(triphenylphosphoranylidene)acetate (740 mg, 2.21 mmol) in dichloromethane (30 mL) was added dropwise over 30 min. Stirring was continued for a further 10 min. Analysis by TLC (50 : 50, ethyl acetate–hexane) indicated disappearance of the oxo-compound and the formation of a new product of higher Rf value. The solution was allowed warm to room temperature and was washed with water (20 mL), the organic layer was separated, dried and concentrated under reduced pressure to leave crude as a brown oil. Purification by chromatography (silica gel, hexane–ethyl acetate, 70 : 30) yielded the benzhydryl ester Wittig product benzhydryl-7-[(Z)-(methoxycarbonyl)methylidene)]cephalosporanate (19)—a yellow–green glassy gel (485.7 mg, 0.984 mmol, 45% yield)—as a mixture of Δ2 and Δ3 isomers (approx. 70 : 30): For characterisation purposes (see below) a small sample of each isomer was obtained by chromatography (silica gel, hexane–ethyl acetate, 70 : 30); for the subsequent sulfoxide formation and reduction—required to obtain a larger sample of the Δ3 isomer—the isomer mixture of 19 was used. Δ2 isomer: δH (270 MHz, CDCl3) 1.96 (s, 3H, –C(O)CH3), 3.82 (s, 3H, CH3O), 4.57 (d, J = 12.62 Hz, 1H, H-3′A), 4.64 (d, J = 12.62 Hz, 1H, H-3′B), 5.24 (d, J = 1.48 Hz, 1H, H-4), 5.74 (d, J = 1.48 Hz, 1H, H-6), 6.33 (d, J = 0.99 Hz, 1H, H-2), 6.48 (d, J = 0.99 Hz, 1H, C(O)CH), 6.91 (s, 1H, CH(Ar)2), 7.25–7.38 (m, 10H, ArH); δC (67.93 MHz, CDCl3) 20.75 (–C(O)CH3), 51.40, 52.64, 53.67, 65.76 (C-4, CH3O, C-6, C-3′), 79.28 (CH(Ar)2), 114.99 119.15, 124.26 (C-3, C-2, C(O)CH), 126.85, 127.19, 128.40, 128.54, 128.76, 128.84, (C Ar), 138.95, 138.99 (C Ar), 155.03 (C-7), 158.10, 164.21, 166.46, 170.52 (4 × CO). See below for characterisation data on the Δ3 isomer.
Sulfoxide formation
The Δ2/Δ3 mixture, (485.7 mg, 0.98 mmol) was dissolved in dichloromethane (29 ml) and the solution was cooled to 0 °C. m-Chloroperoxybenzoic acid, 86% (256.6 mg, 1.49 mmol) was dissolved in dichloromethane (4 mL) and was added dropwise to the cold cephalosporin solution. The solution was stirred at 0 °C for 40 min and reaction progress was monitored by TLC. A cold solution of 10% sodium sulfite (7.1 mL) and 0.1 M sodium hydrogen carbonate (7.1 mL) was added and the biphasic mixture was stirred at 0 °C for 10 min. The layers were separated. The organic layer was washed with 0.1 M sodium hydrogen carbonate (3 × 7 mL) and 20% NaCl solution (10 mL), dried over magnesium sulfate and concentrated under vacuum to yield the benzhydryl sulfoxide ester 20—a yellow solid (349.2 mg, 0.685 mmol, 70% yield) as a mixture of α/β-sulfoxide isomers. For characterisation purposes (see below) a small sample of each isomer was obtained by chromatography (silica gel, ethyl acetate–hexane, 80 : 20); for the subsequent reduction process the isomer mixture of 20 was used. β-Isomer (20) (less polar material):17 found C, 61.27; H, 4.59; N, 2.60%. C26H23NO8S requires C, 61.29; H, 4.55; N, 2.75%; δH (270 MHz, CDCl3) 2.08 (s, 3H, –C(O)CH3), 3.55 (d, J = 16.49 Hz, 1H, H-2A), 3.84 (d, J = 16.49 Hz, 1H, H-2B), 3.87 (s, 3H, CH3O), 4.99 (d, J = 14.65 Hz, 1H, H-3′A), 5.16 (d, J = 14.65 Hz, 1H, H-3′B), 5.26 (d, J = 1.47 Hz, 1H, H-6), 6.62 (d, J = 1.47 Hz, 1H, C(O)CH), 6.97 (s, 1H, CH(Ar)2), 7.25–7.50 (m, 10H, ArH); δC (67.93 MHz, CDCl3) 20.66 (–C(O)CH3), 51.15, 52.97, 62.00 (C-2, CH3O, C-3′, C-6), 80.10 (CH(Ar)2), 119.82 (C(O)CH), 126.86, 127.24, 127.36, 128.31, 128.34, 128.58, 128.66, (C-3, C-4,C Ar), 131.48, 138.84 (C Ar), 148.41 (C-7), 157.12, 159.33, 163.40, 170.23 (4 × CO). α-Isomer (20) (more polar material): found C, 60.90; H, 4.53; N, 2.55%. C26H23NO8S requires C, 61.29; H, 4.55; N, 2.75%; δH (270 MHz, CDCl3) 2.02 (s, 3H, –C(O)CH3), 3.30 (d, J = 19.42 Hz, 1H, H-2A), 3.84 (d, J = 19.42 Hz, 1H, H-2B), 3.85 (s, 3H, CH3O), 4.69 (d, J = 13.92 Hz, 1H, H-3′A), 5.21 (d, J = 13.92 Hz, 1H, H-3′B), 5.18 (d, J = 1.47 Hz, 1H, H-6), 6.63 (d, J = 1.47 Hz, 1H, C(O)CH), 7.00 (s, 1H, CH(Ar)2), 7.26–7.50 (m, 10H, ArH).
Benzhydryl-7-[(Z)-(methoxycarbonyl)methylidene)]cephalosporanate (19, Δ3 isomer) by sulfoxide reduction
The α/β-isomer mixture of 20 (182.4 mg, 0.358 mmol) was dissolved in DMF (8 ml) and cooled to −40 °C. Phosphorus trichloride (63 µL, 0.72 mmol) was added. The mixture was allowed to warm from −40 to −10 °C while stirring under nitrogen. The reaction mixture was added to cold water (21 mL) and the product 19 (Δ3 isomer) precipitated out immediately. The precipitate, a yellow solid, was collected by vacuum filtration, washed with water and dried over P2O5 for 3 days, (142.2 mg, 0.288 mmol, 80%). Found C, 63.15; H, 4.61; N, 2.67%. C26H23NO7S requires C, 63.28; H, 4.70; N, 2.84%; νmax (KBr)/cm−1 1781, 1732; δH (300 MHz, CDCl3) 2.02 (s, 3H, –C(O)CH3), 3.39 (d, J = 18.60 Hz, 1H, H-2A), 3.62 (d, J = 18.60 Hz, 1H, H-2B), 3.84 (s, 3H, CH3O), 4.76 (d, J = 13.51 Hz, 1H, H-3′A), 4.98 (d, J = 13.51 Hz, 1H, H-3′B), 5.49 (d, J = 1.20 Hz, 1H, H-6), 6.48 (d, J = 1.20 Hz, 1H, C(O)CH), 6.99 (s, 1H, CH(Ar)2), 7.22–7.48 (m, 10H, ArH).Sodium-7-(Z)-methoxycarbonylmethylidenecephalosporanate (19′)
Removal of the benzhydryl ester of 19 (Δ3 isomer), (86 mg, 0.174 mmol), was effected as described above for 7′ above. The salt 19′ was obtained as a pale yellow solid (40.1 mg, 0.115 mmol, 66%). Free acid: ESI-HRMS for C13H13NO7S: [M–H]−1 calcd: 326.0033, found: 326.0319; νmax (KBr)/cm−1 3440, 1784, 1773, 1723; δH (270 MHz, CDCl3) 2.12 (s, 3H, –C(O)CH3), 3.45 (d, J = 18.80 Hz, 1H, H-2A), 3.67 (d, J = 18.80 Hz, 1H, H-2B), 3.85 (s, 3H, CH3O), 4.96 (d, J = 13.85 Hz, 1H, H-3′A), 5.15 (d, J = 13.85 Hz, 1H, H-3′B), 5.52 (s, 1H, H-6), 6.50 (s, 1H, C(O)CH); δC (67.93 MHz, CDCl3), δC 20.80(–C(O)CH3), 27.86, 52.67, 57.85, 63.27 (C-2, C-6, CH3O, C-3′), 80.10 (CH(Ar)2), 117.99 (C(O)CH), 126.24, 126.90, 151.90 (C-3, C-4, C-7), 158.09, 163.16, 163.91, 171.20 (4 × CO). Sodium salt 19′: δH (270 MHz, D2O) 2.11 (s, 3H, –C(O)CH3), 3.43 (d, J = 18.3 Hz, 1H, H-2A), 3.73 (d, J = 18.3 Hz, 1H, H-2B), 3.84 (s, 3H, CH3O), 4.74 (d, J = 12.6 Hz, 1H, H-3′A), 4.89 (d, J = 12.6 Hz, 1H, H-3′B), 5.65 (s, 1H, H-6), 6.58 (s, 1H, C(O)CH).
Sodium-7-(Z)-methoxycarbonylmethylidenecephalosporanate sulfoxide (β-isomer) (20′)
Removal of the benzhydryl ester of 20 (β-isomer), (137.4 mg, 0.270 mmol), was effected as described above for 7′ above. For the free acid: δH (270 MHz, CDCl3) 2.13 (s, 3H, –C(O)CH3), 3.64 (d, J = 16.58 Hz, 1H, H-2A), 3.88 (s, 3H, CH3O), 3.98 (d, J = 16.58 Hz, 1H, H-2B), 5.03 (d, J = 14.60 Hz, 1H, H-3′A), 5.21 (d, J = 14.35 Hz, 1H, H-3′B), 5.39 (d, J = 1.24 Hz, 1H, H-6), 6.62 (d, J = 1.48 Hz, 1H, C(O)CH). The salt 20′ was obtained as a pale yellow solid (42.2 mg, 0.115 mmol, 43%). δH (270 MHz, D2O) 2.12 (s, 3H, –C(O)CH3), 3.82 (d, J = 16.34 Hz, 1H, H-2A), 3.83 (s, 3H, CH3O), 4.23 (d, J = 16.34 Hz, 1H, H-2B), 5.61 (d, J = 1.22 Hz, 1H, H-6), 6.74 (d, J = 1.22 Hz, 1H, C(O)CH).Sodium-7-(Z)-methoxycarbonylmethylidenedesacetoxycephalosporanate (21′)
Benzhydryl-7-(Z)-methoxycarboylmethylidenedesacetoxycephalosporanate (21) was prepared, starting from commercially available 7-aminodesacetoxycephalosporanic acid, by the methodology given above for 19. The benzhydryl ester (367 mg, 50%) was obtained as a mixture of Δ2 and Δ3 isomers (37 : 63) which were separated by chromatography. Δ3 isomer: mp 58–60 °C; ESI-HRMS for C24H22NO5S: [M + H]+1 calcd: 436.1219, found: 436.1235; νmax (KBr)/cm−1 1781 (lactam), 1732; δH (270 MHz, CDCl3) 2.12 (s, 3H, CH3), 3.23 (d, J = 18.06 Hz, 1H, H-2A), 3.51 (d, J = 18.06 Hz, 1H, H-2B), 3.83 (s, 3H, OCH3), 5.48 (d, J = 1.73 Hz, 1H, H-6), 6.44 (d, J = 1.24 Hz, 1H, C(O)CH) 6.96 (s, 1H, CH(Ar)2), 7.21–7.51 (m, 10H, ArH); δC (67.93 MHz, CDCl3), 20.22 (CH3), 31.69 (C-2), 52.49 (OCH3), 57.96 (C-6), 79.20 (CH(Ar)2), 116.36 (C(O)CH), 123.80, 127.14, 127.55, 128.02, 128.06, 128.47, 128.56 (C-3, C Ar), 133.23, 139.52, 139.64 (C-4, C Ar), 153.59 (C-7), 157.75, 161.32, 164.21 (3 × CO).
This ester was treated with aluminium chloride as described above for 7′ to remove the benzhydryl group and give the free acid of 21′ (40 mg, 0.149 mmol, 69%): ESI-HRMS for C11H11NO5S: [M–H]−1 calcd: 268.0280, found: 268.0292; δH (270 MHz, CDCl3) 2.26 (s, 3H, CH3), 3.29 (d, J = 18.56 Hz, 1H, H-2A), 3.62 (d, J = 18.56 Hz, 1H, H-2B), 3.84 (s, 3H, OCH3), 4.83 (br s, 1H, OH), 5.51 (d, J = 0.62 Hz, 1H, H-6), 6.46 (d, J = 0.99 Hz, 1H, C(O)CH). The free acid (30.3 mg, 0.113 mmol) was dissolved in ethyl acetate and extracted with aqueous sodium hydrogen carbonate (8 mg, 0.096 mmol) and the resulting aqueous layer was lyophilised to leave 21′ as a bright yellow solid (22 mg, 0.076 mmol, 79%); νmax (KBr)/cm−1 1746 (lactam), 1704; δH (270 MHz, D2O) 1.95 (s, 3H, CH3), 3.27 (d, J = 18.31 Hz, 1H, H-2A), 3.70 (d, J = 18.06 Hz, 1H, H-2B), 3.81 (s, 3H, OCH3), 5.61 (s, 1H, H-6), 6.52 (s, 1H, C(O)CH).
Fluorescence measurement
Coumarin salt 8′ (0.124 mg, 2.8 × 10−4 mmol) in 0.02 M phosphate buffer (200 µL), pH = 7.2 at 25 °C was combined with varying amounts (0, 6, 30, 60 units) of penicillinase type 1. The fluorescence emission at 448 nm (excitation wavelength 360 nm) was recorded as a function of time on a TECAN, GENios Pro instrument.Kinetic analysis
1H NMR spectra were recorded in D2O buffers (0.02 M phosphate, pH 7.2) at 25 °C. Penicillinase type 1 from B. cereus (Sigma) was used for enzyme-catalysed hydrolysis.2,2-Dimethyl-5-oxo-2,3,5,7a-tetrahydro-pyrrolo[2,1-b]thiazole-3,7-dicarboxylic acid (9′)
See ref. 6 for full characterisation (1H, 13C NMR spectra and ESI-HRMS).2,2,6-Trimethyl-5-oxo-2,3,5,7a-tetrahydro-pyrrolo[2,1-b]thiazole-3,7-dicarboxylic acid (9″)
Following completion of the kinetic analysis run of the β-lactamase-catalysed hydrolysis of 7′: δH (270 MHz, D2O) 1.56 (s, 3H, α-CH3), 1.57 (s, 3H, β-CH3), 2.02 (d, J = 1.5 Hz, 3H, CH3), 4.16 (s, 1H, H-3), 5.90 (s, 1H, H-8). After lyophilisation: ESI-HRMS for C11H13NO5S: [M–H]−1 calcd: 270.0436, found: 270.0432.6-(Z)-Carboxymethylidenepenicillanic acid (11)
Following completion (12 h) of the buffer-catalysed hydrolysis of 8′: δH (270 MHz, D2O) 1.48 (s, 3H, α-CH3), 1.54 (s, 3H, β-CH3), 4.28 (s, 1H, H-3), 5.92 (s, 1H, H-5), 6.31 (s, 1H, C(O)CHC, sidechain). After lyophilisation: ESI-HRMS for C10H11NO5S: [M–H]−1 calcd: 256.0280, found: 256.0285.
3-Methyl-6-oxo-6,8a-dihydro-2H-pyrrolo[2,1-b][1,3]thiazine-4,8-dicarboxylic acid (22)
Following completion of the kinetic analysis run of the β-lactamase-catalysed hydrolysis of 21′: δH (270 MHz, D2O) 1.95 (s, 3H, CH3), 3.29 (d, J = 17.81 Hz, 1H, H-2A), 3.73 (d, J = 17.81 Hz, 1H, H-2B), 5.31 (s, 1H, H-9), 6.60 (s, 1H, H-7). Distilled water (20 mL) was added, the solution was acidified to pH 2.2 and extracted with ethyl acetate (2 × 20 mL). The organic layer was dried and concentrated under reduced pressure to leave the free acid of 22 as a yellow solid: ESI-HRMS for C10H9NO5S: [M–H]−1 calcd 254.0123 found 254.0130; δH (270 MHz, (CD3)2CO) 2.06 (s, 3H, CH3), 2.96 (br s, 1H, OH), 3.40 (d, J = 17.81 Hz, 1H, H-2A), 3.84 (d, J = 17.81 Hz, 1H, H-2B), 5.48 (d, J = 0.87 Hz, 1H, H-9), 6.92 (d, J = 1.24 Hz, 1H, H-7).2,2-dimethyl-5-oxo-2,3,5,6-tetrahydropyrrolo[2,1-b]thiazole-3,7-dicarboxylic acid 7-methyl ester (23)
The benzhydryl ester 10 was treated as described above for 7′. The free acid of 23 was obtained as a yellow glassy solid (49 mg, 0.181 mmol, 66%); mp 51–52 °C (dec); ESI-HRMS for C11H13NO5S: [M–H]−1 calcd 270.0436 found 270.0439; νmax (KBr)/cm−1 3438, 1733, 1687; δH (270 MHz, CDCl3) 1.65 (s, 3H, α-CH3), 1.76 (s, 3H, β-CH3), 3.55 (d, J = 23.26 Hz, 1H, H-6A), 3.65 (d, J = 23.26 Hz, 1H, H-6B), 3.75 (s, 3H, OCH3), 4.42 (s, 1H, H-3), 6.00 (br s, 1H, OH); δC (67.93 MHz, CDCl3), 25.30 (α-CH3), 32.99 (β-CH3), 40.27 (CH2), 51.52 (CH3O), 62.41, 65.00 (C-2, C-3), 97.21 (C-7), 156.27 (C-8), 164.20, 169.32, 173.31 (3 × CO). The free acid (31 mg, 0.114 mmol) was dissolved in ethyl acetate and extracted with aqueous sodium hydrogen carbonate (9.1 mg, 0.108 mmol) and the resulting aqueous layer was lyophilised to leave as a dark green solid (27.4 mg, 0.093 mmol, 82%): δH (270 MHz, D2O) 1.59 (s, 3H, α-CH3), 1.72 (s, 3H, β-CH3), 3.64 (s, 2H, CH2), 3.76 (s, 3H, OCH3), 4.29 (s, 1H, H-3).
Acknowledgements
We are grateful to R. O'Dwyer of the University of Limerick for preparation of the β-lactamase-producing E. coli culture and for assistance with the fluorescence measurements, and to C. J. Schofield, B. Lienard and W. Sobey of the University of Oxford, UK, and to L. Horsfall and J.-M. Frère of the Université de Liège, Belgium for the antibiotic and β-lactamase inhibition assays. We are grateful to D. Rai of the CSCB, University College Dublin for the HRMS, and to IRCSET for financial support (C.C.R.).References
- T. A. Murphy, L. E. Catto, S. E. Halford, A. T. Hadfield, W. Minor, T. R. Walsh and J. Spencer, J. Mol. Biol., 2006, 357, 890–903 CrossRef CAS and references therein.
- (a) C. H. O'Callaghan, R. B. Sykes and S. E. Staniforth, Antimicrob. Agents Chemother., 1976, 245–248 CAS; (b) D. Greenwood and F. O'Grady, Antimicrob. Agents Chemother., 1976, 249–252 CAS; (c) S. Mobashery and M. Johnson, J. Biol. Chem., 1986, 261, 7879–7887 CAS; (d) S. Mobashery and M. Johnson, Biochemistry, 1987, 26, 5878–5884 CrossRef CAS; (e) T. P. Smyth, M. E. O'Donnell, M. J. O'Connor and J. O. St Ledger, Tetrahedron, 2000, 56, 5699–5707 CrossRef CAS and references therein.
- (a) H. A. Albrecht, G. Beskid, K.-K. Chan, J. G. Christenson, R. Cleeland, K. H. Deitcher, N. H. Georgopapadakou, D. D. Keith, D. L. Pruess, J. Sepinwall, A. C. Specian, R. L. Then, M. Weigele, K. F. West and R. Yang, J. Med. Chem., 1990, 33, 77–86 CrossRef CAS; (b) H. A. Albrecht, G. Beskid, J. G. Christenson, J. W. Durkin, V. Fallat, N. H. Georgopapadakou, D. D. Keith, F. M. Konzelmann, E. R. Lipschitz, D. H. McGarry, J.-A. Siebelist, C. C. Wei, M. Weigele and R. Yang, J. Med. Chem., 1991, 34, 669–675 CrossRef CAS; (c) H. A. Albrecht, G. Beskid, J. G. Christenson, N. H. Georgopapaakou, D. D. Keith, F. M. Konzelmann, D. L. Pruess, P. L. Rossman and C. C. Wei, J. Med. Chem., 1991, 34, 2857–2864 CrossRef CAS; (d) H. A. Albrecht, G. Beskid, J. G. Christenson, K. H. Dietcher, N. H. Georgopapadakou, D. D. Keith, F. M. Konzelmann, D. L. Pruess and C. C. Wei, J. Med. Chem., 1994, 37, 400–407 CrossRef CAS; (e) T. P. Demuth, R. E. White, R. A. Tietjen, R. J. Storrin, J. R. Skuster, J. A. Andersen, C. C. McOsker, R. Freedman and F. J. Rourke, J. Antibiot., 1991, 44, 200–209 CAS; (f) P. M. Hershberger and T. P. Demuth, Adv. Exp. Med. Biol., 1998, 456, 239–267 CAS and references therein.
- An interesting exploitation of this elimination reaction pattern has been reported in the development of a carbapenem antibiotic: H. Rosen, R. Hajdu, L. Silver, H. Kropp, K. Dorso, J. Kohler, J. G. Sundelof, J. Huber, G. G. Hammond, J. J. Jackson, C. J. Gill, R. Thompson, B. A. Pelak, J. H. Epstein-Toney, G. Lankas, R. R. Wilkening, K. J. Wildonger, T. A. Blizzard, F. P. DiNinno, R. W. Ratcliffe, J. V. Heck, J. W. Kozarich and M. L. Hammond, Science, 1999, 283, 703–706 Search PubMed.
- (a) T. P. Smyth, M. E. O'Donnell, M. J. O'Connor and J. O. St, Ledger, J. Org. Chem., 1998, 63, 7600–7618 CrossRef CAS; (b) T. P. Smyth, M. J. O'Connor and M. E. O'Donnell, J. Org. Chem., 1999, 64, 3132–3138 CrossRef CAS; (c) T. P. Smyth, M. E. O'Donnell, M. J. O'Connor and J. O. St, Ledger, Tetrahedron, 2000, 56, 5699–5707 CrossRef CAS.
- C. C. Ruddle and T. P. Smyth, Chem. Commun., 2004, 2332–2333 RSC.
- (a) A. J. Kirby, in Advances in Physical Organic Chemistry, vol. 17, ch. 3, 1980 Search PubMed; (b) L. Mandolini, in Advances in Physical Organic Chemistry, vol. 22, ch. 1, 1986 Search PubMed.
- J. W. Grant and T. P. Smyth, J. Org. Chem., 2004, 69, 7965–7970 CrossRef CAS.
- (a) J. D. Buynak, B. Geng, B. Bachmann and L. Hua, Bioorg. Med. Chem. Lett., 1995, 5, 1513–1518 CrossRef CAS; (b) J. D. Buynak, A. S. Rao and S. Nidamarthy, Tetrahedron Lett., 1998, 39, 4945–4946 CrossRef CAS; (c) J. D. Buynak, WO Pat. 99/33837, 1999; (d) J. D. Buynak, K. Wu, B. Bachmann, D. Khasnis, L. Hua, H. K. Nguyen and C. L. Carver, J. Med. Chem., 1995, 38, 1022–1034 CrossRef CAS; (e) J. D. Buynak, V. R. Doppalapudi and G. Adam, Bioorg. Med. Chem. Lett., 2000, 10, 853–857 CrossRef CAS.
- See data in the ESI†.
- (a) N. Tsuji, K. Nagashima, M. Kobayashi, J. Shoji, T. Kato, Y. Terui, H. Nakai and M. Shiro, J. Antibiot., 1982, 35, 24–31 CAS; (b) Y. Kimura, K. Motokawa, H. Nagata, Y. Kameda, S. Matsuura, M. Mayama and T. Yoshida, J. Antibiot., 1982, 35, 32–38 CAS; (c) K. Murakami, M. Doi and T. Yoshida, J. Antibiot., 1982, 35, 38–45.
- One unit hydrolyzes 1.0 µmole of benzylpenicillin per min at pH 7.0 at 25 °C.
- The E. coli strain Top10 (Invitrogen) containing the plasmid pIGXH, expressing an ampicillin resistant gene, was used. This material was prepared by R. O'Dwyer in the Department of Chemical and Environmental Sciences, University of Limerick.
- See ref. 10 in: B. E. Maryanoff, M. J. Constanzo, S. O. Nortey, M. N. Greco, R. P. Shank, J. J. Schupsky, M. P. Ortegon and J. L. Vaught, J. Med. Chem., 1998, 41, 1315–1343 Search PubMed.
- (a) T. Ritter, K. Stanek, I. Larrosa and E. M. Carreira, E. M., Org. Lett., 2004, 6, 1513–1514 CrossRef CAS; (b) A. F. Barrero, J. E. Oltra, M. Alvarez and A. Rosales, J. Org. Chem., 2002, 67, 5461–5469 CrossRef CAS.
- The screening assays were carried out at the Chemistry Research Laboratory, University of Oxford, UK, and at the Centre d'Ingénierie des Proteines, Université de Liège, Belgium, under the supervision of Prof. C. J. Schofield, and Prof. J.-M. Frère, respectively.
- For details of sulfoxide isomer assignment see: J. B. Doherty, et al., J. Med. Chem., 1990, 33, 2513–2521 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectral sequences for reaction of 7′ and 20′ with β-lactamase enzyme; Gaussian energy values for addition of methoxide to the α-face of an ethylidene β-lactam unit. See DOI: 10.1039/b614758e |
| This journal is © The Royal Society of Chemistry 2007 |