A new synthetic approach to biaryls of the rhazinilam type. Application to synthesis of three novel phenylpyridine-carbamate analogues
Received
15th September 2006
, Accepted 18th October 2006
First published on 23rd November 2006
Abstract
The synthesis of three novel racemic phenylpyridine-carbamate analogues of rhazinilam and their biological evaluation as inhibitors of microtubule assembly and disassembly by interaction with tubulin are described. The sterically hindered ortho-disubstituted biaryl unit as the challenging key structural element is first obtained by a sequential regiocontrolled nucleophilic addition of a lithium ortho-lithiohomobenzylic alkoxide species to 3-bromo-5-oxazolyl pyridine as the electrophile and a subsequent oxidation step. The incorporation of the amino group by replacement of the bromide has been achieved using a Buchwald–Hartwig amination coupling. Ultimate deprotection steps furnished free-amino and free-hydroxyl appendages which were connected by phosgenation to furnish the nine-membered median carbamate ring.
Introduction
(−)-Rhazinilam 1 is a tetracyclic phenyl-pyrrole alkaloid isolated from various Apocynaceae.1 It was found both to induce in vitro spiralization of microtubules (vinblastin effect) and to inhibit the cold-induced disassembly of microtubules1 (paclitaxel effect). As a consequence of these unique antitubulin properties, (−)-rhazinilam showed significant in vitro cytotoxicity towards various cancer cell lines but no activity was found in vivo.1 Thus structure–activity relationships on rhazinilam analogues are of considerable interest.1 Recently three-dimensional quantitative structure–activity relationships (3D-QSAR) from all available analogues were investigated.2 The biphenyl carbamate analogue (−)-2 mimicking the structure of (−)-rhazinilam developed by Guéritte et al.3 is currently the most active analogue with a 2-fold activity on microtubule disassembly compared to 1 and a similar cytotoxicity. The synthesis of several biphenyl analogues allowed the establishment of the main structural elements for maximum antitubulin activity which are the presence of the biaryl unit bridged by a nine-membered carbamate ring (ring B) as well as a quaternary center mimicking the stereogenic center of 1. Our laboratory proposed in 2001 the first phenylpyridine analogues of rhazinilam4 and their biological evaluation confirmed that the replacement of the lactam by a carbamate function enhances the antitubulin activity. The best biological result was obtained with rac-3 which is however six times less active on the inhibition of microtubule disassembly than (−)-rhazinilam. In continuation of this study, we embarked on a program aimed at the synthesis of new phenylpyridine analogues derived from biphenyl analogue 2 by replacing the aniline moiety (ring A) by an aminopyridine system and keeping the nine-membered carbamate ring and a quaternary center as essential structural elements for the antitubulin activity. We expected thus to evaluate the effect of the charge distribution on the aniline moiety of (−)-rhazinilam (ring A) on the antitubulin activity. Here we wish to report the synthesis of three novel racemic phenylpyridine-carbamate analogues of rhazinilam 4a, 4b and 4c (Fig. 1) and their biological evaluation.
Results ans discussion
Our planned approach to phenylpyridine-carbamates 4 bearing the nitrogen atom on the aniline moiety (ring A) is shown in Scheme 1. The nine-membered carbamate ring could be installed by ring-closure of the aminoalcohol 5 (route a). The final ring-closure could also be achieved by an intramolecular palladium catalyzed Buchwald–Hartwig amidation of bromocarbamate 7 obtained by carbamoylation of the bromoalcohol 6 (route b). The two ortho-substituted biaryl precursors 5, 6 could be obtained a priori using cross-coupling protocols but Guéritte and Baudoin have pointed out the sensitivity of cross-couplings toward steric hindrance in biphenyl series due in particular to the presence of the highly hindered quaternary center.3c Thus, in order to prepare phenylpyridine analogue rac-3,4 Rocca et al. opted for a post-alkylation of the picolinic site to install the quaternary carbon after formation of the biaryl unit. However this elegant approach cannot be applied to the preparation of the two biaryl precursors 5 and 6 due to the much lower acidity of the benzylic site compared to the picolinic one. In this paper we envisage the formation of the pivotal ortho-substituted biaryl unit using a key nucleophilic addition of the lithium ortho-lithioarylalkoxide salt 9 to an adequate activated bromopyridine scaffold 8, followed by an oxidative step to regenerate the pyridine nucleus.
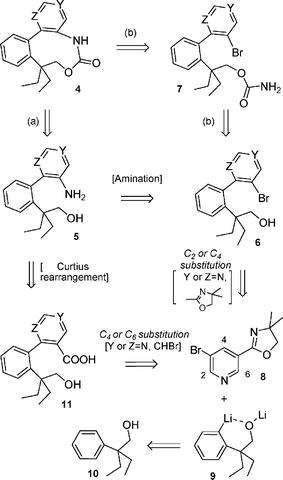 |
| | Scheme 1 | |
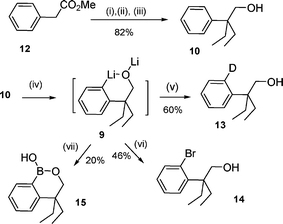
We initially investigated the unreported ortho-lithiation of the α,α’-ethylhomobenzylic alcohol 10 ready prepared from methyl 2-phenyl acetate 12 in 82% yield using a double ethylation and reduction sequence depicted in Scheme 2. The α,α’-ethylhomobenzylic alcohol 10 was subjected to a series of strong bases. The lithiation yield was determined by measuring the ratios of starting material and deuterated product (10 : 13) after an external D2O quench technique (Table 1). We first applied Meyer and Seebach's conditions reported for the lithiation of α,α’-methylbenzylic alcohol,5 similar to homobenzylalcohol 10 (Table 1-entry 1). After quenching with D2O, a mixture of 10 and 13 was obtained in a 4 : 1 ratio. We turned to stronger bases, sec-BuLi and tert-BuLi. The lithiation was carried out on a broad range of temperatures and using different equivalents of base. After several experiments summarized in Table 1 (entries 2–7), the best result was obtained with the treatment of 10 with 3 equiv. of sec-BuLi in hexane at 70 °C for 6 h, which provided a 1 : 9 mixture of 10–13 in 60% yield after chromatography (entry 6). The ortho-bromo α,α’-ethylhomobenzylic alcohols 14 could be prepared in modest 46% yields using 1,2-dibromo-tetrachloroethane as a halogenating agent. The borylation of the lithium ortho-lithioarylalkoxide salt 9 could also be performed with trimethylborate as an electrophile followed by hydrolysis with HCl to give the first prepared six-membered ring oxaborine 15 in 20% yield over two steps.
Table 1 Lithiation–D2O trapping of 10
| Entry |
Base |
Equiv. |
t/°C |
Time/h |
Ratio 10–13x–ya(%)b |
|
NMR conversion to deuterated compound.
Isolated yield obtained using the optimal metallation conditions.
|
| 1 |
n-BuLi–TMEDA |
2 |
70 |
12 |
4 : 1 (—) |
| 2 |
sec-BuLi |
2 |
70 |
6 |
7 : 3 (—) |
| 3 |
sec-BuLi |
3 |
r.t. |
24 |
6 : 4 (—) |
| 4 |
sec-BuLi |
5 |
r.t. |
24 |
7 : 3 (—) |
| 5 |
sec-BuLi |
3 |
40 |
32 |
3 : 7 (50) |
|
6
|
sec-BuLi
|
3
|
70
|
6
|
1 : 9 (60)
|
| 7 |
tert-BuLi |
3 |
r.t. |
32 |
3 : 7 (55) |
The 3-bromo-5-oxazolyl pyridine scaffold 8 was prepared on a multigram-scale from commercially available 3-bromo-5-cyano pyridine 16 by treatment with 2-amino-2-methyl propanol 17 in the presence of ZnCl2 as a catalyst6 (Scheme 3). Although Ottow and co-workers have previously reported the nucleophilic addition of 3-anisylmagnesium bromide to 5-bromo-3-pyridine carboxamide7 similar to our scaffold 8, no previous report has given sound support to the nucleophilic addition of aryllithium species to protected 3-bromo-5-carboxypyridine derivatives. Such a nucleophilic addition was explored by reacting 3-bromo-5-oxazolyl pyridine 8 with phenyllithium at room temperature in THF for 1 h as a typical experiment.8 Subsequent oxidative treatment of the crude reaction mixture with chloranil in refluxing toluene for 2 h afforded the expected phenylpyridine 18 in good 87% isolated yield (Scheme 3). The latter showed two characteristic singlets corresponding to the H2 and H6 protons in the 1H NMR spectrum indicating that the phenyl group was exclusively introduced to the C4 site of the pyridine nucleus in agreement with Ottow's observations.7 We next investigated the addition of lithium ortho-lithioarylalkoxide salt 9 to 3-bromo-5-oxazolyl pyridine 8. After generation from 10 as previously described, 9 was added to 3-bromo-5-oxazolyl pyridine 8 in THF at room temperature for 1 h.
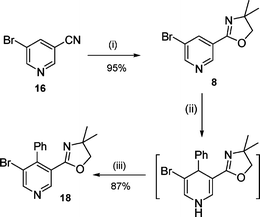 |
| | Scheme 3
Reagents and conditions: (i) 2-amino-2-methylpropan-1-ol 17, ZnCl2 (10%mol), PhCl, Δ, 2 days; (ii) PhLi (1 equiv.), THF, r.t., 1 h then NH4Cl; (iii) chloranil, toluene, Δ, 2 h. | |
Treatment of the crude reaction with chloranil in refluxing toluene for 2 h provided a biaryl compound in 26% overall yield. Careful analysis surprisingly revealed the introduction of the phenyl moiety at the C2 site of the pyridine nucleus leading to bromoalcohol 6a (Scheme 4).
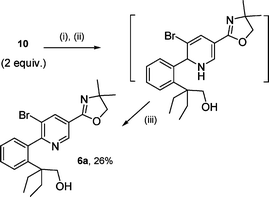 |
| | Scheme 4
Reagents and conditions: (i) sec-BuLi (6 equiv.), hexane, 70 °C, 6 h; (ii) 8 (1 equiv.), THF, r.t., 1 h then NH4Cl; (iii) chloranil, toluene, Δ, 2 h. | |
Meyers, Hauck, Knaus and Bourguignon et al. previously demonstrated that the regiochemical outcome of the nucleophilic addition of aryllithium species to 3-oxazolylpyridine may occur indifferently at C2, C4 and C6 sites depending mainly on three factors: polarity of the solvent, addition temperature and steric hindrance of the nucleophilic reagent.8 Unfortunately we were not able to direct the nucleophilic addition of lithio anion 9 at another position whatever the solvent (Et2O and toluene were used), the temperature or the use of TMEDA. Construction of the nine-membered cyclic carbamate from bromoalcohol 6a was next investigated. To this purpose carbamate 7a was first prepared in 70% yield via carbamoylation of 6a in the presence of trichloroacetylisocyanate9 (Scheme 5) before cyclisation of the N-metallated derivative of 7a. Thus, 7a was N-metallated by treatment with sodium hydride in DMF at room temperature10 (Table 2-entry 1). However the expected annelated product 4a was not formed, but instead the seven-membered-ring biaryl-ether 19 was exclusively obtained in excellent 90% yield. We could hypothesize that the competitive elimination of isocyanic acid11 from the N-metallated carbamate produced in situ the lithium oxanion which then substituted the bromine atom in an intramolecular way. We then turned to the transition metal-catalyzed carbamatation. Ring-closure via Buchwald's amidation by using Cu(I) and a diamine ligand as catalyst12 (Table 2-entry 2) led to the cleavage of the carbamoyl function affording bromoalcohol 6a in 70% yield. The Pd(0)-catalyzed intramolecular carbamatation was then tested using two specific ligands, P(tBu)313 and Xantphos14 (Table 2-entries 3 and 4). In both cases no expected annelated compound rac-4a could be obtained. Similar to previous observations, decarbamoylation occurred, especially when using Xantphos as a ligand (Table 2-entry 4), and the resulting bromoalcohol 6a was then partially converted in low yields to the seven membered-ring biaryl-ether 19.
Table 2 Assays of nine-membered ring-closing of 7a
| Entry |
Conditions |
%-4aa |
%-6aa |
%-7aa |
%-19a |
|
NMR conversion except for entry 1.
Yield of isolated product.
|
| 1 |
NaH, DMF, r.t., 12 h |
None |
0 |
0 |
90b |
| 2 |
CuI, (MeNHCH2)2, K2CO3, toluene, Δ, 16 h |
None |
70 |
30 |
0 |
| 3 |
Pd(dba)2, P(tBu)3HBF4, NaOPh, toluene, Δ,16 h |
None |
16 |
60 |
24 |
| 4 |
Pd2(dba)3, Xantphos, Cs2CO3, dioxane, 16 h |
None |
60 |
20 |
20 |
At this stage it appeared necessary to protect the free-alcohol function of 6a before performing the substitution of the bromine by an amino group. The TBS-alcohol protection of 6a was effected using a standard procedure15 to give 21 in 67% yield. As Akiba and co-workers have previously shown that N-silyldihydropyridine could be readily reoxidized to pyridine through treatment with chloranil,16 the TBS-alcohol protection was directly performed after the nucleophilic addition of lithium ortho-lithioarylalkoxide salt 9 to scaffold 8via subsequent addition of tert-butyldimethylsilylchloride (TBSCl). Without isolation, protected N-silyldihydropyridine intermediate 20 (Scheme 6) was then subjected to oxidative treatment with chloranil giving rise to 21 in 21% yield through a one-pot four step synthesis. A classical palladium-catalyzed Buchwald–Hartwig amination17 of 21 provided then the bis-protected biarylic aminoalcohol 22 in excellent 83% yield (Scheme 6). Next, both N-PMB and O-TBS deprotections were carried out with TFA18 and the resulting crude free-amino ester 23 thus obtained was immediately hydrolyzed by simple basic treatment with K2CO3 in a mixture of MeOH–H2O to give biarylic aminoalcohol 5a in 78% yield over two steps (Scheme 6). The final internal carbamatation was first attempted by treating 5a with triphosgene.4 Surprisingly this process failed due probably to the poor nucleophilicity of the amino group in the presence of the electron-withdrawing oxazolyl group on the pyridine nucleus. Nucleophilic activation of both free-amino and free-alcohol groups remained unsuccessful after metallation of 5a with 2.5 equivalents of n-BuLi and treatment of the resulting N,O-dilithio anions with diethylcarbonate19 at room temperature for 16 h. Finally, the direct phosgenation of 5a in the presence of NEt320 was achieved providing the desired phenylpyridine carbamate rac-4a in an excellent 84% yield (Scheme 6). The corresponding ethyl ester rac-4b and carboxylic acid rac-4c were obtained via H2SO4-induced cleavage21 of the oxazolyl group in EtOH or in H2O respectively but in low 20 and 15% yields due to the undesired hydrolysis of the carbamate group (Scheme 7). Milder methods for oxazoline hydrolysis involving the prior formation of oxazolinium salts22 and subsequent treatment with NaOH couldn't be applied as the treatment of rac-4a with CH3I exclusively afforded the N-methylpyridinium salt 24. Treatment of rac-4a with NaOCl also remained unsuccessful leading only to the N-chlorocarbamate analogue 25 (Scheme 7).
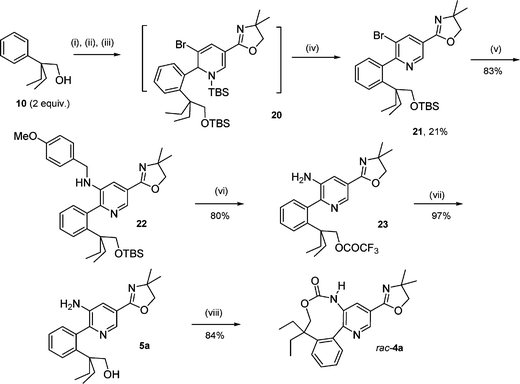 |
| | Scheme 6
Reagents and conditions: (i) sec-BuLi (6 equiv.), THF, 70 °C, 6 h; (ii) 8 (1 equiv.), THF, r.t., 1 h; (iii) TBSCl, r.t., 16 h; (iv) chloranil, toluene, Δ, 2 h; (v) p-methoxybenzylamine (1.2 equiv.), Pd2dba3, BINAP, tBuONa, toluene, Δ, 16 h; (vi) TFA, r.t., 16 h; (vii) K2CO3, MeOH–H2O, r.t., 1 h; (viii) phosgene (1 equiv.), NEt3, THF, 0 °C to 25 °C, 1 h. | |
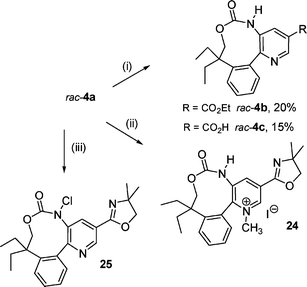 |
| | Scheme 7
Reagents and conditions: (i) H2SO4–EtOH or H2O (1 : 9), Δ, 5 h; (ii) CH3I, CH3CN, r.t., 12 h; (iii) NaOCl, Bu4NHSO4, AcOEt, r.t., 4 h. | |
The cytotoxicity and the antitubulin activity of the new phenylpyridine analogues 4a, 4b and 4c were evaluated and compared to those of (−)-1 (Table 3). The seven-membered-ring bridged biaryl-ether 19 was also submitted to the same test as the ring structure analogue of the antimitotic (−)-colchicine. Phenylpyridine analogues 4a, 4b and 4c were weakly active in the polymerization and depolymerization of microtubules. In addition, no cytotoxicity was found on KB cancer cells as well as on human breast adeno-carcinoma MCF7. It was previously demonstrated that the addition of sterically hindered substituents on the aniline moiety (ring A) of (−)-2a lowers the interaction with tubulin.1 This suggests that the inactivity of rac-4a, rac-4b and rac-4c is imputable to the substitution of the pyridine nucleus rather than to its electron-deficient character. The biological evaluation of the unsubstituted pyridine analogue would be necessary to verify this assumption. The seven-membered-ring biaryl-ether 19 was respectively 15 and 50 times less active than (−)-1 in tubulin polymerization and depolymerization and no cytotoxicity towards adeno-carcinoma MCF7 as well as KB cancer cells was observed.
Table 3 Cytotoxicity and antitubulin activity of (−)-rhazinilam, rac-4a–c and 19
| Compound |
Cytotox. KB cell line IC50/µMa |
Cytotox. MCF7 cell line IC50/µMa |
Inhibition of microtubule disassembly IC50/µMb |
Inhibition of microtubule assembly IC50/µMb |
|
IC50 is the concentration of compound corresponding to 50% growth inhibition after 72 h incubation.
IC50 is the concentration of compound required to inhibit 50% of the rate of microtubule assembly or disassembly (in = inactive).
Percentage of inhibition of assembly or disassembly at 10 mg L−1 (IC50 could not be measured).
|
|
1
|
0.6 |
4.0 |
3.7 |
6.7 |
|
4a
|
in |
in |
1%c |
32%c |
|
4b
|
in |
in |
12%c |
18%c |
|
4c
|
in |
in |
in |
in |
|
19
|
in |
in |
190 |
110 |
Conclusion
The hindered rhazinilam-like phenylpyridine carbamates were prepared for the first time by using a one-pot three step sequence with the generation of the lithium ortho-lithioarylalkoxide salt 9 by ortho-lithiation, subsequent nucleophilic addition to the 3-bromo-5-oxazolyl pyridine scaffold 8 at the C2 position and a final oxidative step. The method was directly applied to the preparation of three novel phenyl-3-carboxypyridyl-carbamate analogues 4a, 4b and 4c of rhazinilam as racemic mixtures in 11, 2.2 and 1.6% overall yields respectively. These new analogues proved to be inactive in the inhibition of the polymerization and depolymerization of tubulin. Other substituted as well as unsubstituted phenylpyridine carbamates will be synthesized by extension of the nucleophilic addition of the lithium ortho-lithioarylalkoxide salt 9 to other activated pyridine scaffolds.
Experimental
General
Melting points were measured on a Kofler apparatus. NMR spectra were recorded on a Bruker AM 300 spectrometer with residual protic solvent as the internal reference. Chemical shifts are quoted in ppm and coupling constants in Hz. IR spectra were recorded using KBr cells on a Perkin-Elmer FT IR 205 spectrometer. Elemental analyses were performed on a Carlo Erba 1106 apparatus. High-resolution mass spectra (HRMS) were recorded by the department service. THF was distilled from benzophenone–Na. Silica gel (Geduran Si 60, 0.063–0.200 mm) was purchased from Merck. The compounds 12 and 17, the solutions of n-BuLi, sec-BuLi and tert-BuLi and the ligands P(tBu3)HBF4, Xantphos were used as received.
2-Ethyl-2-phenylbutan-1-ol (10).
A solution of n-BuLi (48 ml of 2.5 M solution in hexane, 120 mmol) was slowly added, at −78 °C under N2, to a stirred solution of 2,2′-bipyridyl (4 mg, 0.02 mmol) and N-diisopropylamine (DIPEA) (16.8 ml, 120 mmol) in THF (120 ml). The mixture was stirred for 15 min at −78 °C and a solution of methyl 2-phenylacetate 12 (4.31 ml, 30 mmol) in THF (50 ml) was added. After an additional 30 min stirring period, EtI (12 ml, 150 mmol) was added. The mixture was allowed to warm to r.t. for 3 h and H2O (100 ml) was added. The product was extracted with CH2Cl2 (3 × 70 ml) and the combined organic extracts were dried (MgSO4), filtered and concentrated under vacuo. The residue was dissolved in THF (50 ml) and the above deprotonation–ethylation sequence was repeated (LDA (60 mmol) in THF (60 ml) and EtI (6 ml, 75 mmol)). The residue was purified by flash chromatography (Cy–EA 98 : 2) to give 2-methyl-2-ethylphenylbutanoate (5.7 g, 92%) as a pale yellow oil. To a solution of 2-methyl-2-ethylphenylbutanoate (500 mg, 2.4 mmol) in THF (10 ml) was slowly added a solution of LiAlH4 (91 mg, 2.4 mmol) in THF (5 ml) at 0 °C. The mixture was stirred at 25 °C for 2 h under N2 and cooled at 0 °C before H2O (91 µl), NaOH (2 M, 91 µl) and H2O (270 µl) were successively added. After filtration though a short pad of celite, the product was extracted with AcOEt (3 × 10 ml). The combined organic extract was dried (MgSO4), filtered and concentrated under vacuo. The residue was purified by distillation (112 °C, 2 mbar) to give 10 (384 mg, 90%) as a colorless oil (Found: C, 80.79; H, 10.23. Calc. for C12H18O: C, 80.85; H, 10.18%); IR νmax/cm−1 3307 (OH), 2969; 1H NMR δH (300 MHz, CDCl3): 0.75 (6 H, t, J 7.2, 2 × CH2CH3), 1.07 (1 H, s, OH), 1.74 (4 H, m, 2 × CH2CH3), 3.74 (2 H, d, J 6.0, CH2OH), 7.22 (1 H, m, 4-H), 7.33 (4 H, m, Ph); 1H NMR δH (300 MHz, DMSO): 0.59 (6 H, t, J 7.1, 2 × CH2CH3), 1.64 (4 H, m, 2 × CH2CH3), 3.61 (2 H, d, J 4.9, CH2OH), 4.48 (1 H, t, J 4.9, OH), 7.12 (1 H, m, 4-H), 7.26 (4 H, m, Ph); 13C NMR δC (75 MHz, CDCl3): 7.9 (2 × CH3), 26.0 (2 × CH2), 46.0 (C(Et)2), 67.1 (CH2OH), 125.9 (CHar), 126.9 (2 × CHar), 128.3 (2 × CHar), 144.4 (CAr).
2-(2-Deuteriophenyl)-2-ethyl-butan-1-ol (13).
A solution of sec-BuLi (2.6 ml of 1.3 M solution in cyclohexane–hexane 98 : 2, 3.36 mmol) was slowly added, at r.t. under N2, to a stirred solution of 2-ethyl-2-phenylbutan-1-ol (10, 200 mg, 1.12 mmol) in hexane (2 ml). The mixture was refluxed for 6 h and cooled to 0 °C before D2O (0.6 ml, 30 mmol) was added. The temperature was slowly allowed to warm to r.t. and water (2 ml) was added. The product was extracted with AcOEt (3 × 5 ml) and the combined organic extract was washed with brine, dried (MgSO4), filtered and concentrated under vacuo. The residue was purified by flash chromatography (Cy–EA 8 : 2) to give 13 (125 mg, 60%) as a colorless oil. 1H NMR δH (300 MHz, DMSO): 0.59 (6 H, t, J 7.1, 2 × CH2CH3), 1.64 (4 H, m, 2 × CH2CH3), 3.61 (2 H, d, J 4.9, CH2OH), 4.48 (1 H, t, J 4.9, OH), 7.12 (1 H, m, 4-H), 7.26 (3 H, m, Ph).
2-(2-Bromophenyl)-2-ethyl-butan-1-ol (14).
A solution of sec-BuLi (2.6 ml of 1.3 M solution in cyclohexane–hexane 98 : 2, 3.36 mmol) was slowly added, at r.t. under N2, to a stirred solution of 2-ethyl-2-phenylbutan-1-ol (10, 200 mg, 1.12 mmol) in hexane (2 ml). The mixture was refluxed for 6 h and cooled to 0 °C before a solution of 1,2-dibromotetrachloroethane (1.82 g, 5.6 mmol) in THF (3 ml) was added. The temperature was allowed to warm to 25 °C and the mixture was stirred for 16 h. Water (2 ml) was added and the product was extracted with AcOEt (3 × 5 ml). The combined organic extract was washed with brine, dried (MgSO4), filtered and concentrated under vacuo. The residue was purified by flash chromatography (Cy–EA 8 : 2) to give 14 (134 mg, 46%) as a colorless oil (Found: C, 56.34; H, 6.65. Calc. for C12H17BrO: C, 56.04; H, 6.66%); IR νmax/cm−1 3380, 2965; 1H NMR δH (300 MHz, CDCl3): 0.71 (6 H, t, J 7.2, 2 × CH2CH3), 1.20 (1 H, t, J 6.0, OH), 1.98 (2 H, m, CH2CH3), 2.10 (2 H, m, CH2CH3), 4.08 (2 H, d, J 6.0, CH2OH), 7.05 (1 H, t, J 6.8, 4-H), 7.26 (2 H, m, 5-H, 6-H), 7.60 (1 H, d, J 7.5, 3-H); 13C NMR δC (75 MHz, CDCl3): 8.9 (2 × CH3), 25.3 (2 × CH2), 48.5 (C(Et)2), 64.7 (CH2OH), 122.8 (Car), 127.5 (CHar), 128.2 (CHar), 131.4 (CHar), 136.4 (CHar), 142.1 (Car).
3,4-Dihydro-1-hydroxy-4,4-diethyl-(2,1)-benzoxaborine (15).
A solution of sec-BuLi (2.6 ml of 1.3 M solution in cyclohexane–hexane 98 : 2, 3.36 mmol) was slowly added, at r.t. under N2, to a stirred solution of 2-ethyl-2-phenylbutan-1-ol (10, 200 mg, 1.12 mmol) in hexane (2 ml). The mixture was refluxed for 6 h and cooled to −78 °C before trimethyl borate (1.35 ml, 12.00 mmol) was added. The solution was allowed to warm to 25 °C and the mixture was continued for 16 h. Saturated NH4Cl (15 ml) was added. The separated aqueous extract was then acidified with HCl (3 M) to pH = 1, extracted with AcOEt (3 × 10 ml) and the combined organic extract was washed with NaOH (2 N, 3 × 10 ml). The aqueous extract was acidified with HCl (3 M) to pH = 5 and extracted with AcOEt (3 × 15 ml). The combined organic extract was dried (MgSO4), filtered and concentrated under vacuo to give 15 (46 mg, 20%) as a white solid without further purification; mp 145 °C (Found: C, 70.47; H, 8.52. Calc. for C12H17BO2: C, 70.63; H, 8.40%); IR νmax/cm−1 3215 (OH), 1459, 805 (B–C); 1H NMR δH (300 MHz, DMSO): 0.71 (6 H, t, J 7.5, 2 × CH2CH3), 1.55–1.66 (4 H, m, 2 × CH2CH3), 3.90 (2 H, s, CH2OB), 7.22 (2 H, m, Ph), 7.43 (1 H, t, J 7.2, Ph), 7.69 (1 H, d, J 7.5, Ph), 8.42 (1 H, s, OH); 13C NMR δC (75 MHz, CDCl3): 8.6 (2 × CH3), 29.0 (2 × CH2), 43.0 (C(Et)2), 71.5 (CH2O), 125.1 (CHar), 125.8 (CHar), 131.2 (CHar), 133.6 (CHar), 151.3 (Car).
3-Bromo-5-(4,5-dihydro-4,4-dimethyloxazol-2-yl)pyridine (8).
To a suspension of zinc chloride (371 mg, 2.73 mmol) in chlorobenzene (55 mL) was added, at r.t. under N2, 5-bromonicotinonitrile (5 g, 27.3 mmol) and 2-amino-2-methylpropan-1-ol (17, 2.74 mL, 28.6 mmol). The resulting mixture was refluxed for 48 h and solvent was removed under vacuo. The residue was dissolved in CH2Cl2 (30 mL) and the resulting organic phase was washed with H2O (30 mL). The separated aqueous phase was extracted with CH2Cl2 (3 × 10 mL) and the combined organic extract was dried (MgSO4), filtered and concentrated under vacuo. The residue was purified by flash chromatography (EA) to give 8 (6.61 g, 95%) as white powder; mp 91 °C (Found: C, 47.14; H, 4.33; N, 10.83. Calc for C10H11BrN2O: C, 47.08; H, 4.35; N, 10.98%); IR νmax/cm−1 3033, 2870, 1935, 1651; 1H NMR δH (300 MHz, CDCl3): 1.39 (6 H, s, 2 × CH3), 4.15 (2 H, s, CH2), 8.38 (1 H, dd, J 1.9 and 2.2, 4-H), 8.74 (1 H, d, J 2.2, 2-H), 9.02 (1 H, d, 1H, J 1.9, 6-H); 13C NMR δC (75 MHz, CDCl3): 28.7 (2 × CH3), 69.0 (C(Me)2), 80.1 (CH2), 120.5 (Car), 125.8 (Car), 138.1 (CHar), 147.5 (CHar), 152.0 (CHar), 159.1 (CN).
3-Bromo-5-(4,5-dihydro-4,4-dimethyloxazol-2-yl)-4-phenylpyridine (18).
PhLi (0.39 ml of 2 M solution in Bu2O, 0.78 mmol) was slowly added, at r.t. under N2, to a solution of 3-bromo-5-(4,5-dihydro-4,4-dimethyloxazol-2-yl)pyridine 8 (200 mg, 0.78 mmol) in THF (6 ml). The solution was stirred at r.t. for 1 h. Saturated NH4Cl (10 ml) was added and the mixture was extracted with CH2Cl2 (3 × 10 ml). The combined organic layers were dried (MgSO4), filtered and concentrated in vacuo. The crude residue was then dissolved in toluene (30 ml) and subsequently treated with chloranil (193 mg, 0.78 mmol). The mixture was refluxed for an additional 2 h and washed successively with aqueous NaOH (12%, 3 × 30 ml) and water (30 ml). The separated organic layer was dried (MgSO4), filtered and concentrated under vacuo. The crude residue was purified by flash chromatography (P–EA 3 : 7) to give 18 (224 mg, 87%) as a yellow oil (Found: C, 58.29; H, 8.07; N, 4.62. Calc. for C16H15BrN2O: C, 58.02; H, 8.46; N, 4.56%); IR νmax/cm−1 2964, 1651; 1H NMR δH (300 MHz, CDCl3): 1.03 (6 H, s, 2 × CH3), 3.56 (2 H, s, CH2), 7.13 (2 H, m, Ph), 7.27 (3 H, m, Ph), 8.64 (1 H, s, Py), 8.72 (1 H, s, Py); 13C NMR δC (75 MHz, CDCl3): 28.3 (2 × CH3), 68.3 (C(Me)2), 79.9 (CH2), 122.6 (CAr), 127.1 (CAr), 128.6 (CHAr), 128.8 (CHAr), 129.0 (CAr), 137.2 (CHAr), 149.0 (CHAr), 149.7 (CAr), 153.9 (CHAr), 160.4 (CN).
2-(2-(3-Bromo-5-(4,5-dihydro-4,4-dimethyloxazol-2-yl)pyridin-2-yl)phenyl)-2-ethylbutan-1-ol (6a).
A solution of sec-BuLi (7.75 ml of 1.3 M solution in cyclohexane–hexane 98 : 2, 10.11 mmol) was added dropwise, at r.t. under N2, to a stirred solution of 2-ethyl-2-phenylbutan-1-ol (10, 600 mg, 3.37 mmol) in hexane (6 ml). The mixture was refluxed for 6 h, cooled to 25 °C and a solution of 3-bromo-5-(4,5-dihydro-4,4-dimethyloxazol-2-yl)pyridine (8, 430 mg, 1.69 mmol) in THF (6 ml) was added. The resulting mixture was stirred at 25 °C for 1 h before saturated NH4Cl (10 ml) was added. The product was extracted with AcOEt (3 × 10 ml) and the combined organic extract was dried (MgSO4), filtered and concentrated in vacuo. The residue was dissolved in toluene (34 ml) and chloranil (416 mg, 1.69 mmol) was added. The mixture was refluxed for an additional 2 h. The cooled solution was washed with NaOH (12%, 3 × 30 ml) and water (30 ml) and the separated organic phase was dried (MgSO4), filtered and concentrated under vacuo. The residue was purified by flash chromatography (DCM–Ac 9 : 1) to give 6a (190 mg, 26%) as a brown solid; mp 46 °C (Found: C, 61.27; H, 6.61; N, 6.10. Calc. for C22H27BrN2O2: C, 61.26; H, 6.31; N, 6.49%); IR νmax/cm−1 3364 (OH), 1651; 1H NMR δH (300 MHz, CDCl3): 0.64 (3 H, t, J 7.5, CH2CH3), 0.88 (3 H, t, J 7.2, CH2CH3), 1.25 (2 H, m, CH2CH3), 1.42 (6 H, s, 2 × CH3), 1.74 (1 H, m, CH2CH3), 1.83 (1 H, m, CH2CH3), 3.16 (1 H, d, J 10.9, CH2OH), 3.31 (1 H, s, OH), 3.46 (1 H, d, J 10.9, CH2OH), 4.18 (2 H, s, CH2), 6.99 (1 H, d, J 7.5, 6-H), 7.28 (1 H, t, J 7.5, 5-H), 7.43 (1 H, t, J 8.3, 4-H), 7.55 (1 H, d, J 8.3, 3-H), 8.57 (1 H, d, J 1.7, 4′-H), 8.99 (1 H, d, J 1.7, 6′-H); 13C NMR δC (75 MHz, CDCl3): 8.8 (2 × CH3), 27.3 (CH2), 27.6 (CH2), 28.7 (2 × CH3), 48.9 (C(Et)2), 67.7 (CH2OH), 68.4 (C(Me)2), 79.8 (CH2), 121.9 (Car), 124.8 (Car), 126.0 (CHar), 129.0 (CHar), 130.2 (CHar), 131.7 (CHar), 139.4 (Car), 140.7 (CHar), 141.9 (Car), 146.3 (CHar), 159.1 (CN), 164.6 (Car).
2-(2-(3-Bromo-5-(4,5-dihydro-4,4-dimethyloxazol-2-yl)pyridin-2-yl)phenyl)-2-ethylbutyl carbamate (7a).
Trichloroacetyl isocyanate (101 µl, 0.85 mmol) was slowly added, at 0 °C under N2, to a stirred solution of compound 6a (280 mg, 0.65 mmol) in freshly dry CH2Cl2 (6 ml). The mixture was allowed to warm to room temperature and stirred for an additional 30 min. After removal of the solvent under vacuo, the crude product obtained was dissolved in methanol (6 ml) and the resulting solution was treated with potassium carbonate (196 mg, 1.42 mmol). The mixture was stirred for 3 h and water (10 ml) was then added. The mixture was extracted with ethyl acetate (3 × 15 ml) and the combined organic layers were dried (MgSO4), filtered and concentrated under vacuo. The crude material was purified by flash chromatography (DCM–Ac 9 : 1) to give 7a (216 mg, 70%) as an orange solid; mp 70 °C (Found: C, 58.16; H, 6.02; N, 8.93. Calc. for C23H28BrN3O3: C, 58.23; H, 5.95; N, 8.86%); IR νmax/cm−1 3338 (NH2), 1725; 1H NMR δH (300 MHz, CDCl3): 0.72 (6 H, m, 2 × CH2CH3), 1.42 (6 H, s, 2 × CH3), 1.53 (4 H, m, 2 × CH2CH3), 4.03 (1 H, d, J 10.9, CH2OCO), 4.15 (1 H, d, J 10.9, CH2OCO), 4.17 (2 H, s, CH2), 4.56 (2 H, s, NH2), 7.01 (1 H, dd, J 7.5 and 1.5, 6-H), 7.28 (1 H, t, J 7.5, 5-H), 7.37–7.47 (2 H, m, 3-H, 4-H), 8.52 (1 H, d, J 1.9, 4′-H), 9.03 (1 H, d, J 1.9, 6′-H); 13C NMR δC (75 MHz, CDCl3): 8.9 (CH3), 9.0 (CH3), 28.0 (CH2), 28.4 (CH2), 28.7 (2 × CH3), 47.2 (C(Et)2), 68.1 (CH2OCO), 68.4 (C(Me)2), 79.8 (CH2), 122.0 (Car), 124.5 (Car), 126.1 (CHar), 128.7 (CHar), 129.3 (CHar), 131.8 (CHar), 139.7 (Car), 140.2 (CHar), 141.7 (Car), 146.5 (CHar), 157.2 (CO), 159.3 (CN), 164.7 (Car).
7,7-Diethyl-6,7-dihydro-4,5-dihydro-3-(4,4-dimethyloxazol-2-yl)benzoxepino[1,2,b]pyridine (19).
To a stirred solution of compound 7a (70 mg, 0.15 mmol) in DMF (1 ml) were added small portions of NaH (13 mg of 60% in mineral oil, 0.32 mmol). The resulting mixture was allowed to warm to 100 °C for 16 h and saturated NH4Cl (2 ml) was added. The product was extracted with AcOEt (3 × 5 ml). The combined organic extract was dried (MgSO4), filtered and concentrated under vacuo. The crude material was purified by flash chromatography (DCM–Ac 9 : 1) to give 19 (46 mg, 90%) as a yellow oil. IR νmax/cm−1 2968, 1652; 1H NMR δH (300 MHz, CDCl3): 0.76 (6 H, m, 2 × CH2CH3), 1.40 (6 H, s, 2 × CH3), 1.67 (2 H, m, CH2CH3), 1.89 (2 H, m, CH2CH3), 4.13 (2 H, s, CH2), 4.26 (2 H, s, CH2OAr), 7.38 (3 H, m, Ph), 7.80 (1 H, d, J 1.9, 4-H), 8.59 (1 H, m, Ph), 8.91 (1 H, d, J 1.9, 6-H); 13C NMR δC (75 MHz, CDCl3): 9.0 (2 × CH3), 28.8 (2 × CH3), 31.9 (2 × CH2), 48.3 (C(Et)2), 68.2 (C(Me)2), 79.6 (CH2OAr), 79.7 (CH2), 123.7 (Car), 126.9 (CHar), 127.2 (CHar), 128.4 (CHar), 129.0 (CHar), 132.4 (CHar), 136.3 (Car), 143.2 (CHar), 145.1 (Car), 149.0 (Car), 155.1 (Car), 160.3 (CN); HRMS (IC+, tert-butane) calcd. for C22H27N2O2 [(M + H)+] 351.2072, found 351.2065.
3-Bromo-5-(4,5-dihydro-4,4-dimethyloxazol-2-yl)-2-(2-(3-(((1,1-dimethylethyl)dimethylsilyloxy)methyl)pentan-3-yl)phenyl)pyridine (21).
Method A (from compound 6a).
Tert-butylchlorodimethylsilane (82 mg, 0.55 mmol) was added to a stirred solution of compound 6a (214 mg, 0.50 mmol) and 1H-imidazole (54 mg, 0.80 mmol) in DMF (1 ml). The resulting mixture was stirred at 25 °C under N2 for 2 h and water (10 ml) was added. The product was extracted with CH2Cl2 (3 × 15 ml) and the combined organic extract was dried (MgSO4), filtered and concentrated under vacuo. The residue was purified by flash chromatography (DCM–Ac 9.5 : 0.5) to give 21 (183 mg, 67%) as an orange oil. IR νmax/cm−1 2961, 1653, 1085; 1H NMR δH (300 MHz, CDCl3): −0.14 (3 H, s, MeSi), −0.12 (3 H, s, MeSi), 0.64–0.70 (6 H, m, 2 × CH2CH3), 0.80 (9 H, s, (CH3)3Si), 1.42 (6 H, s, 2 × CH3), 1.45–1.71 (4 H, m, 2 × CH2CH3), 3.36 (1 H, d, J 9.2, CH2OSi), 3.44 (1 H, d, J 9.2, CH2OSi), 4.17 (2 H, s, CH2), 6.99 (1 H, dd, J 7.5 and 1.3, 3′-H), 7.22 (1 H, td, J 8.3 and 1.3, 4′-H), 7.35–7.44 (2 H, m, 5′-H, 6′-H), 8.51 (1 H, d, J 1.9, 4-H), 9.03 (1 H, d, J 1.9, 6-H); 13C NMR δC (75 MHz, CDCl3): −5.4 (MeSi), −5.3 (MeSi), 9.2 (CH3), 9.3 (CH3), 18.5 (CMe3), 26.1 (3 × CH3), 28.7 (2 × CH3), 29.1 (CH2), 30.1 (CH2), 49.4 (C(Et)2), 64.1 (CH2OSi), 68.4 (C(Me)2), 79.8 (CH2), 122.0 (Car), 124.4 (Car), 125.6 (CHar), 128.5 (CHar), 129.6 (CHar), 131.2 (CHar), 139.8 (Car), 139.9 (CHar), 142.4 (Car), 146.3 (CHar), 159.3 (CN), 165.1 (Car); HRMS (DIC+, tert-butane) calcd. for C28H42N2O2SiBr [(M + H)+] 545.2199–547.2183, found 545.2206–547.2174.
Method B (one-pot procedure from compound 10).
A solution of sec-BuLi (1.25 ml of 1.3 M solution in cyclohexane–hexane 98 : 2, 1.68 mmol) was slowly added, at r.t. under N2, to a stirred solution of 2-ethyl-2-phenylbutan-1-ol (10, 200 mg, 0.56 mmol) in hexane (1 ml). The mixture was refluxed for 6 h, cooled to 25 °C and a solution of 3-bromo-5-(4,5-dihydro-4,4-dimethyloxazol-2-yl)pyridine (8, 72 mg, 0.28 mmol) in THF (1 ml) was added. The resulting mixture was stirred at r.t. for 1 h before a solution of tert-butylchlorodimethylsilane (253 mg, 1.68 mmol) in THF (1 ml) was added. The mixture was stirred for 16 h, water (2 ml) was added and the product was extracted with AcOEt (3 × 5 ml). The combined organic extract was dried (MgSO4), filtered and concentrated in vacuo. The residue was dissolved in toluene (7 ml) and chloranil (69 mg, 0.28 mmol) was added. The mixture was refluxed for an additional 2 h and the cooled solution was washed with NaOH (12%, 3 × 5 ml) and water (5 ml). The separated organic phase was dried (MgSO4), filtered and concentrated under vacuo. The residue was purified by flash chromatography (DCM–Ac 9.5 : 0.5) to afford 21 (30 mg, 21%) as an orange oil with the same characteristic analysis as above.
N-(4-Methoxybenzyl)-5-(4,5-dihydro-4,4-dimethyloxazol-2-yl)-2-(2-(3-(((1,1-dimethylethyl)dimethylsilyloxy)methyl)pentan-3-yl)phenyl)pyridin-3-amine (22).
To a stirred solution of compound 21 (300 mg, 0.55 mmol) in dry toluene pre-degassed by argon bubbling for 30 min (3 ml) were added 4-methoxybenzylamine (87 µl, 0.66 mmol), sodium tert-butoxide (73 mg, 0.76 mmol), BINAP (14 mg, 0.02 mmol) and Pd2dba3 (8 mg, 0.008 mmol). The resulting mixture was refluxed for 16 h under N2. After filtration through a short pad of celite, the solvents were removed under vacuo and the residue was purified by flash chromatography (DCM–Ac 9 : 1) to give 22 (274 mg, 83%) as a yellow oil. IR νmax/cm−1 3416 (NH), 1651, 1084; 1H NMR δH (300 MHz, CDCl3): −0.11 (6 H, s, 2 × MeSi), 0.57–0.67 (6 H, m, 2 × CH2CH3), 0.80 (9 H, s, (CH3)3Si), 1.40 (6 H, s, 2 × CH3), 1.47–1.61 (2 H, m, CH2CH3), 1.69–1.74 (2 H, m, CH2CH3), 3.45 (2 H, s, CH2OSi), 3.71 (1H, m, NH), 3.77 (3 H, s, OMe), 4.12 (2 H, s, CH2), 4.16–4.32 (2 H, m, CH2N), 6.82 (2 H, d, J 8.6, H-PhOMe), 7.03 (1 H, dd, J 7.3 and 1.3, 3′-H), 7.14 (2 H, d, J 8.6, 2 × H-PhOMe), 7.24 (1 H, t, J 7.1, 4′-H), 7.34 (1 H, td, J 7.1 and 1.7, 5′-H), 7.40–7.45 (2 H, m, 4-H, 6′-H), 8.51 (1 H, d, J 1.7, 6-H); 13C NMR δC (75 MHz, CDCl3): −5.3 (2 × MeSi), 9.1 (CH3), 9.2 (CH3), 18.5 (CMe3), 26.1 (3 × CH3), 28.7 (2 × CH3), 29.1 (CH2), 29.6 (CH2), 47.5 (CH2N), 49.4 (C(Et)2), 55.5 (OCH3), 63.6 (CH2OSi), 68.0 (C(Me)2), 79.4 (CH2), 114.3 (CHar), 115.3 (CHar), 124.0 (Car), 126.6 (CHar), 128.6 (CHar), 129.0 (CHar), 130.2 (CHar), 130.4 (Car), 131.2 (CHar), 136.6 (CHar), 137.2 (Car), 142.0 (Car), 144.0 (Car), 152.0 (Car), 159.2 (Car), 161.2 (CN); HRMS (DIC+, tert-butane) calcd. for C36H52N3O3Si [(M + H)+] 602.3778, found 602.3780.
2-(2-(3-Amino-5-(4,5-dihydro-4,4-dimethyloxazol-2-yl)pyridin-2-yl)phenyl)-2-ethylbutyl 2,2,2-trifluoroacetate (23).
A solution of compound 22 (150 mg, 0.25 mmol) in TFA (1.5 ml) was stirred for 16 h at 25 °C. TFA was removed under vacuo and the residue was dissolved in CH2Cl2 (5 ml). Saturated NaHCO3 (5 ml) was added and the separated aqueous phase was extracted with CH2Cl2 (3 × 15 ml). The combined organic extract was dried (MgSO4), filtered and concentrated under vacuo. The residue was purified by flash chromatography (DCM–Ac 9 : 1) to give 23 (93 mg, 80%) as a yellow solid; mp-(degradation). 1H NMR δH (300 MHz, CDCl3): 0.72 (6 H, t, J 7.4, 2 × CH2CH3), 1.41 (6 H, s, 2 × CH3), 1.58–1.78 (4 H, m, 2 × CH2CH3), 3.59 (2 H, s, NH2), 4.15 (2 H, s, CH2), 4.30 (1 H, d, J 11.0, CH2OCO), 4.38 (1 H, d, J 11.0, CH2OCO), 7.08 (1 H, dd, J 7.5 and 1.3, 6-H), 7.33 (1 H, td, J 7.5 and 1.7, 5-H), 7.39–7.46 (2 H, m, 3-H, 4-H), 7.60 (1 H, d, J 1.8, 4′-H), 8.55 (1 H, d, J 1.8, 6′-H); 13C NMR δC (75 MHz, CDCl3): 8.6 (CH3), 8.7 (CH3), 27.6 (CH2), 28.1 (CH2), 28.7 (2 × CH3), 46.9 (C(Et)2), 67.9 (C(Me)2), 70.7 (CH2OCO), 79.7 (CH2), 118.0 (q, JC–F 285, CF3), 121.4 (CHar), 124.1 (Car), 127.5 (CHar), 129.0 (CHar), 129.8 (CHar), 131.7 (CHar), 137.2 (Car), 138.4 (CHar), 140.5 (Car), 141.5 (Car), 150.7 (Car), 159.0 (d, JC–F 41, CO), 161.1 (CN); 19F NMR δF (138 MHz, CDCl3): −75.5 (CF3); m/z (ESI, acetonitrile) 464.1 [(M + H)+].
2-(2-(3-Amino-5-(4,5-dihydro-4,4-dimethyloxazol-2-yl)pyridin-2-yl)phenyl)-2-ethylbutan-1-ol (5a).
To a solution of compound 23 (90 mg, 0.2 mmol) in H2O–CH3OH (1 : 2, 3.5 ml) was added K2CO3 (124 mg, 0.9 mmol). The mixture was stirred at 25 °C for 1 h and the solvents were removed under vacuo. The residue was purified by flash chromatography (DCM–Ac 8 : 2) to give 5a (71 mg, 97%) as a pale yellow solid; mp 65 °C. IR νmax/cm−1 3316–3202, 1649; 1H NMR δH (300 MHz, CDCl3): 0.63 (3 H, t, J 7.3, CH2CH3), 0.87 (3 H, t, J 7.3, CH2CH3), 1.28–1.38 (2 H, m, CH2CH3), 1.40 (6 H, s, 2 × CH3), 1.65–1.85 (2 H, m, CH2CH3), 3.16 (1 H, d, J 12.2, CH2OH), 3.40 (1 H, d, J 12.2, CH2OH), 3.60 (2 H, s, NH2), 4.14 (2 H, s, CH2), 7.04 (1 H, dd, J 7.5 and 1.7, 6-H), 7.29 (1 H, td, J 7.5 and 1.1, 5-H), 7.41 (1 H, td, J 7.5 and 1.7, 4-H), 7.58 (1 H, dd, J 7.5 and 1.1, 3-H), 7.62 (1 H, d, J 1.8, 4′-H), 8.50 (1 H, d, J 1.8, 6′-H); 13C NMR δC (75 MHz, CDCl3): 8.7 (CH3), 8.8 (CH3), 27.5 (CH2), 27.7 (CH2), 28.7 (2 × CH3), 48.8 (C(Et)2), 67.7 (CH2OH), 67.8 (C(Me)2), 79.7 (CH2), 121.6 (CHar), 124.3 (Car), 126.8 (CHar), 129.0 (CHar), 131.0 (CHar), 131.2 (CHar), 136.7 (Car), 138.2 (CHar), 140.4 (Car), 143.6 (Car), 151.4 (Car), 160.9 (CN); HRMS (DIC+, tert-butane) calcd. for C22H30N3O2 [(M + H)+] 367.2260, found 367.2256.
9,9-Diethyl-8,9-dihydro-3-(4,5-dihydro-4,4-dimethyloxazol-2-yl)benzo[f]pyrido[3,2,d]oxazonin-6(5H)-one (rac-4a).
Et3N (162 µl, 1.16 mmol) was added to a stirred solution of compound 5a (170 mg, 0.46 mmol) in THF (8.5 ml) at r.t. under N2. The solution was allowed to cool to 0 °C and phosgene (251 µl of a 20% solution in toluene, 0.47 mmol) was added dropwise. The mixture was stirred at 25 °C for 1 h and saturated NaHCO3 (8 ml) and EtOAc (10 ml) were added. The product was extracted with EtOAc (3 × 10 ml) and the combined organic extract was dried (MgSO4), filtered and concentrated under vacuo. The residue was purified by flash chromatography (DCM–Ac 8 : 2) to give rac-4a (152 mg, 84%) as a pale yellow solid; mp-(decomposition). IR νmax/cm−1 3311, 1743, 1651; 1H NMR δH (300 MHz, CDCl3): 0.70 (3 H, t, J 7.2, CH2CH3), 0.92 (3 H, t, J 7.4, CH2CH3), 1.39 (6 H, s, 2 × CH3), 1.40 (2 H, m, CH2CH3), 1.76 (1 H, m, CH2CH3), 1.90 (1 H, m, CH2CH3), 3.80 (1 H, d, J 10.7, CH2OCO), 4.16 (2 H, s, CH2), 4.21 (1 H, d, J 10.7, CH2OCO), 6.60 (1 H, s, NH), 6.80 (1 H, dd, J 7.5 and 1.7, 3′-H), 7.24 (1 H, t, J 7.4, 4′-H), 7.40 (1 H, td, J 7.4 and 1.7, 5′-H), 7.50 (1 H, d, J 8.1, 6′-H), 7.97 (1 H, d, J 1.9, 4-H), 8.95 (1 H, d, J 1.9, 6-H); 13C NMR δC (75 MHz, CDCl3): 8.3 (CH3), 8.4 (CH3), 24.7 (CH2), 24.8 (CH2), 28.7 (2 × CH3), 48.5 (C(Et)2), 68.3 (C(Me)2), 73.6 (CH2OCO), 79.8 (CH2), 124.0 (Car), 127.0 (CHar), 128.4 (CHar), 129.7 (CHar), 131.3 (CHar), 132.4 (CHar), 133.7 (Car), 139.1 (Car), 141.7 (Car), 145.5 (CHar), 156.4 (Car), 160.1 (CN), 165.4 (CO); HRMS (IC+, tert-butane) calcd. for C23H28N3O3 [(M + H)+] 394.2131, found 394.2134.
9,9-Diethyl-8,9-dihydro-3-(ethoxycarboxy)benzo[f]pyrido[3,2,d]oxazonin-6(5H)-one (rac-4b).
A solution of compound rac-4a (26 mg, 0.07 mmol) in EtOH–conc. H2SO4 (9 : 1, 2 ml) was refluxed for 5 h. After cooling at 25 °C, saturated NaHCO3 was added to pH = 8–9 and the product was extracted with CH2Cl2 (3 × 5 ml). The combined organic extract was dried (MgSO4), filtered and concentrated under vacuo. The residue was purified by flash chromatography (DCM–Ac 9.5 : 0.5) to give rac-4b (5 mg, 20%) as a pale yellow oil. IR νmax/cm−1 3274, 1727; 1H NMR δH (300 MHz, CDCl3): 0.71 (3 H, t, J 7.3, CH2CH3), 0.94 (3 H, t, J 7.3, CH2CH3), 1.44 (5 H, m, CH2CH3, COOCH2CH3), 1.75 (1 H, m, CH2CH3), 1.90 (1 H, m, CH2CH3), 3.82 (1 H, d, J 10.9, CH2OCO), 4.25 (1 H, d, J 10.9, CH2OCO), 4.45 (2 H, q, J 7.1, COOCH2CH3), 6.08 (1 H, s, NH), 6.80 (1 H, dd, J 7.5 and 1.5, 3′-H), 7.26 (1 H, m, 4′-H), 7.43 (1 H, td, J 7.0 and 1.5, 5′-H), 7.54 (1 H, d, J 7.7, 6′-H), 8.05 (1 H, d, J 1.9, 4-H), 9.07 (1 H, d, J 1.9, 6-H); 13C NMR δC (75 MHz, CDCl3): 8.4 (CH3), 8.5 (CH3), 14.7 (CH3Et), 24.9 (2 × CH2), 48.6 (C(Et)2), 62.1 (CH2Et), 73.8 (CH2OCO), 126.0 (Car), 127.1 (CHar), 128.5 (CHar), 129.8 (CHar), 131.1 (CHar), 133.7 (CHar), 133.8 (Car), 139.0 (Car), 141.6 (Car), 147.2 (CHar), 156.1 (Car), 165.2 (CO), 167.2 (CO); HRMS (IC+, tert-butane) calcd. for C21H25N2O4 [(M + H)+] 369.1814, found 369.1794.
3-Carboxy-9,9-diethyl-8,9-dihydrobenzo[f]pyrido[3,2,d]oxazonin-6(5H)-one (rac-4c).
A stirred solution of compound rac-4a (15 mg, 0.04 mmol) in H2SO4 (1.8 N, 2 ml) was refluxed for 5 h. The mixture was allowed to cool to r.t. and saturated NaHCO3 was added to pH = 8–9. The aqueous phase was washed with (1 × 5 ml) and HCl (4 N) was added to pH 4. The separated aqueous phase was extracted with CH2Cl2 (3 × 5 ml) and the combined organic extract was dried (MgSO4), filtered and concentrated under vacuo to give rac-4c (2 mg, 15%) as a pale yellow solid; mp-(decomposition). IR νmax/cm−1 3500–3000, 1722, 1599; 1H NMR δH (300 MHz, CDCl3): 0.72 (3 H, t, J 7.3, CH2CH3), 0.95 (3 H, t, J 7.3, CH2CH3), 1.45 (2 H, m, CH2CH3), 1.77 (1H, m, CH2CH3), 1.93 (1 H, m, CH2CH3), 3.84 (1 H, d, J 10.9, CH2OCO), 4.27 (1 H, d, J 10.8, CH2OCO), 6.80 (2 H, m, NH, 3′H), 7.26 (1 H, m, Ph), 7.44 (1 H, t, J 7.3, Ph), 7.54 (1 H, d, J 8.0, 6′-H), 8.08 (1 H, s, 4-H), 9.09 (1 H, s, 6-H); 13C NMR δC (75 MHz, CD3CD2OD): 8.3 (CH3), 8.5 (CH3), 24.9 (CH2), 25.2 (CH2), 49.2 (C(Et)2), 73.6 (CH2OCO), 127.2 (CHar), 128.8 (CHar), 130.1 (CHar), 132.2 (CHar), 134.3 (CHar), 135.8 (Car), 140.1 (Car), 142.7 (2 × Car), 146.4 (CHar), 157.2 (Car), 165.9 (CN), 169.4 (CO); HRMS (IC−, tert-butane) calcd. for C19H19N2O4 [(M–H)+] 339.1345, found 339.1355.
Acknowledgements
The authors are grateful to Dr F. Guéritte for the support of this work, S. Thoret for tubulin assays and G. Aubert for cytotoxicity assays.
References
- See the recent review of the chemistry and biology of rhazinilam and analogues and references therein: O. Baudoin, D. Guénard and F. Guéritte, Mini-Rev. Org. Chem., 2004, 1, 333–341 Search PubMed.
- H. Morita, K. Awang, A. H. A. Hadi, K. Takeya, H. Itokawa and J. Kobayashi, Bioorg. Med. Chem. Lett., 2005, 15, 1045–1050 CrossRef CAS.
-
(a) C. Pascal, J. Dubois, D. Guénard and F. Guéritte, J. Org. Chem., 1998, 63, 6414–6420 CrossRef CAS;
(b) C. Pascal, J. Dubois, F. Guéritte, D. Guénard, L. Tchertanov, S. Thoret and F. Guéritte, Tetrahedron, 1998, 54, 14737–14756 CrossRef CAS;
(c) O. Baudoin, D. Guénard and F. Guéritte, J. Org. Chem., 2000, 65, 9268–9271 CrossRef CAS;
(d) O. Baudoin, M. Cesario, D. Guénard and F. Guéritte, J. Org. Chem., 2002, 67, 1199–1207 CrossRef CAS;
(e) O. Baudoin, F. Claveau, S. Thoret, A. Herrbach, F. Guénard and F. Guéritte, Bioorg. Med. Chem., 2002, 10, 3395–3400 CrossRef CAS;
(f) A. Herrbach, A. Marinetti, O. Baudoin, D. Guénard and F. Guéritte, J. Org. Chem., 2003, 68, 4897–4905 CrossRef CAS.
- E. Pasquinet, P. Rocca, S. Richalot, F. Guéritte, D. Guénard, A. Godard, F. Marsais and G. Quéguiner, J. Org. Chem., 2001, 66, 2654–2661 CrossRef CAS.
- N. Meyer and D. Seebach, Angew. Chem., Int. Ed. Engl., 1978, 17, 521–522 CrossRef.
- H. Witte and W. Seeliger, Justus Liebigs Ann. Chem., 1974, 996–1009 CrossRef CAS.
-
(a) Ottow has reported the unique example of C4 addition of aryl magnesium bromide to the 5-bromo-3-pyridinecarboxamide: W. Schlecker, A. Huth and E. Ottow, Tetrahedron, 1995, 51, 9531–9542 Search PubMed;
(b) For general examples of the addition of organometallics to pyridinecarboxamides and nicotinates: E. Piers and M. Soucy, Can. J. Chem., 1974, 52, 3563–3564 Search PubMed; K. Akiba, Y. Iseki and M. Wada, Bull. Chem. Soc. Jpn., 1984, 57, 1994–1999;
H. Hans, F. Hoffmann-Emery, G. Rimmler, M. Evans-Roger, H. W. Stahr, P. Wadmeier, EP Pat., 1103546, 2001 CAS; M.-L. Bennasar, C. Juan and J. Bosch, Tetrahedron Lett., 2001, 42, 585–588 CAS; M. L. Bennasar, T. Roca, M. Monerris and C. Juan, Tetrahedron, 2002, 58, 8099–8106 Search PubMed; M.-L. Bennasar, T. Roca and M. Monerris, J. Org. Chem., 2004, 69, 752–756 CrossRef CAS.
- For examples of regiochemical control outcomes of nucleophilic addition of aryllithium to 3-oxazoyl pyridine see: A. I. Meyers and N. R. Natale, Heterocycles, 1982, 18, 13–19 Search PubMed; S. H. Rosenberg and H. Rapoport, J. Org. Chem., 1984, 49, 56–62 CrossRef CAS; A. E. Hauck and C. S. Giam, J. Chem. Soc., Perkin Trans. 1, 1984, 2227–2231 CrossRef CAS; S. K. Dubey and E. E. Knaus, Heterocycles, 1984, 22, 1091–1093 RSC; S. K. Dubey and E. E. Knaus, Heterocycles, 1986, 24, 125–134 CrossRef CAS; P. Binay, G. Dupas, J. Bourguignon and G. Quéguiner, Can. J. Chem., 1987, 65, 648–655 CrossRef CAS.
- D. Dubé, D. Deschênes, J. Tweddell, H. Gagnon and R. Carlini, Tetrahedron Lett., 1995, 36, 1827–1830 CrossRef CAS; M. R. Paleo, N. Aurrecoechea, K.-Y. Jung and H. Rapoport, J. Org. Chem., 2003, 68, 130–138 CrossRef CAS.
- D. Montebugnoli, P. Bravo, E. Corradi, G. Dettori, C. Mioskowski, A. Volonterio, A. Wagner and M. Zanda, Tetrahedron, 2002, 58, 2147–2153 CrossRef CAS; A. J. Bridges and J. P. Sanchez, J. Heterocycl. Chem., 1990, 27, 1527–1536 Search PubMed.
- For previous reports of the elimination of isocyanic acid from N-metallated carbamate see: T. Kaneko, H. Wong and T. W. Doyle, Tetrahedron Lett., 1985, 26, 3923–3926 Search PubMed; H. Al-Rawi and A. Williams, J. Am. Chem. Soc., 1977, 99, 2671–2678 CrossRef CAS.
- A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421–7428 CrossRef CAS.
- J. F. Hartwig, M. Kawatsura, S. I. Hauck, K. H. Shaughnessy and L. M. Alcazar-Roman, J. Org. Chem., 1999, 64, 5575–5580 CrossRef CAS.
- B. H. Yang and S. L. Buchwald, Org. Lett., 1999, 1, 35–37 CrossRef CAS; J. Yin and S. L. Buchwald, Org. Lett., 2000, 2, 1101–1104 CrossRef CAS; A. P. Dishington, P. D. Johnson and J. G. Kettle, Tetrahedron Lett., 2004, 45, 3733–3735 CrossRef CAS.
-
6a was treated with tert-butyldimethylsilylchloride (TBSCl) and imidazole at r.t. for 2 h.
- K. Akiba, Y. Iseki and M. Wada, Bull. Chem. Soc. Jpn., 1984, 57, 1994–1999 CAS.
- S. Wagaw and S. L. Buchwald, J. Org. Chem., 1996, 61, 7240–7241 CrossRef CAS; J. F. Hartwig, Synlett, 1997, 329–340 CrossRef CAS.
- H. Dehmlow, J. D. Aebi, S. Jolidon, Y.-H. Ji, E. M. von der Mark, J. Himber and O. H. Morand, J. Med. Chem., 2003, 46, 3354–3370 CrossRef CAS.
- P. Canonne, R. Boulanger and B. Chantegrel, J. Heterocycl. Chem., 1989, 26, 113–117 CrossRef CAS.
- S. S. Nikam, P.-W. Yuen, B. E. Kornberg, B. Tobias and M. F. Rafferty, J. Org. Chem., 1997, 62, 9331–9334 CrossRef CAS; B. S. Rasmussen, U. Elezcano and T. Skrydstrup, J. Chem. Soc., Perkin Trans. 1, 2002, 14, 1723–1733 RSC.
- M. Reuman and A. I. Meyers, Tetrahedron, 1985, 41, 837–860 CrossRef CAS; J. W. H. Watthey, T. Gavin, M. Desai, B. M. Finn, R. K. Rodebaugh and S. L. Patt, J. Med. Chem., 1983, 26, 1116–1122 CrossRef CAS; R. H. Hutchings and A. I. Meyers, J. Org. Chem., 1996, 61, 1004–1013 CrossRef CAS.
- J. Levin and S. M. Weinreb, Tetrahedron Lett., 1982, 23, 2347–2350 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2007 |
Click here to see how this site uses Cookies. View our privacy policy here.