Cyclic sulfamidates as versatile lactam precursors. An evaluation of synthetic strategies towards (−)-aphanorphine
John F.
Bower
a,
Peter
Szeto
b and
Timothy
Gallagher
*a
aSchool of Chemistry, University of Bristol, Bristol, United Kingdom BS8 1TS. E-mail: t.gallagher@bristol.ac.uk; Fax: 44 117 9298611; Tel: 44 117 9288260
bChemical Development, GlaxoSmithKline, Medicines Research Centre, Stevenage, United Kingdom SG1 2NY
First published on 16th November 2006
Abstract
A full account of studies which led to the efficient asymmetric synthesis of (−)-aphanorphine 1 is reported. Two routes to the key cyclic sulfamidate intermediate 5 are described, the first was based on a chiral auxiliary approach and the second utilised asymmetric hydrogenation methodology. A range of C(3)-substituted lactams (4, 22 and 25) were synthesised and evaluated as precursors for Pd(0) catalysed entries (based on (i) α-arylation of a lactam enolate and (ii) reductive Heck reaction) to the 3-benzazepine core of 1. These approaches were less effective than an aryl radical cyclisation which allowed the completion of a synthesis of 1 in 12 steps from anisaldehyde.
Introduction
(−)-Aphanorphine 1 is a tricyclic alkaloid isolated from the freshwater blue-green alga Aphanizomenon flos-aquae.1 The structural resemblance of 1 to benzomorphan analgesics2 such as pentazocine 2 and eptazocine 3 has led to widespread synthetic attention culminating in several approaches to both racemic and enantiopure 1.3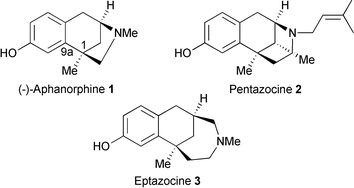
As part of a programme to develop cyclic sulfamidates as N-heterocycle building blocks, we have recently outlined versatile methodologies for the synthesis of a range of enantiopure 5- and 6-ring N-heterocyclic derivatives.4 In particular, the reactions of 1,2- and 1,3-cyclic sulfamidates with functionalised enolates offer a facile and modular entry to α-functionalised pyrrolidinones and piperidinones respectively (Scheme 1).4b,c We have investigated the incorporation of a variety of functionalities at C(3), including esters, phosphonates and sulfides, which then provide a handle for further manipulation and access to other highly functionalised lactam variants.
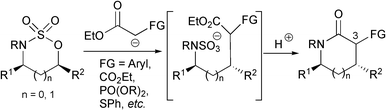 | ||
| Scheme 1 | ||
In order to define further the scope and robustness of this cyclic sulfamidate based methodology we recently reported an efficient asymmetric entry to (−)-aphanorphine 1.5 In this paper we describe full studies directed towards this target molecule and in so doing also provide a further demonstration of the versatility of the heterocyclic strategy that we have developed.
Results and discussion
Our initial approach to (−)-aphanorphine is outlined in Scheme 2 and called for the formation of α-methylated lactam 4 as a precursor for the construction of the core ring system via a palladium-catalysed α-arylation reaction to form the C(1)–C(9a) bond associated with 1. Lactam 4 would in turn be synthesised from cyclic sulfamidate 5 which would be derived from amino acid derivative 6.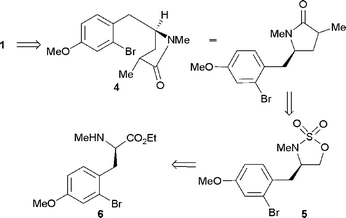 | ||
| Scheme 2 Retrosynthetic analysis of (−)-aphanorphine 1. | ||
Our first generation approach to cyclic sulfamidate 5 is depicted in Scheme 3. Bromination of p-anisaldehyde was achieved in 62% yield in accordance with the procedure described by Durst which employs Comins' methodology for directed ortho-lithiation of aromatic aldehydes.6 A screen of commonly employed electrophilic brominating agents revealed CBr4 to be optimal (Table 1, entry 3).†
| Entrya | Brominating agent | Yieldb |
|---|---|---|
| a All reactions were performed on a 1.00 mmol scale. b Isolated yield (SM = starting material). c 7a and 7b were not separable by chromatography, distillation or crystallisation. | ||
| 1 | Br2 | 18% (+ 70% SM) |
| 2 | BrCH2CH2Br | 6% (+ 81% SM) |
| 3 | CBr4 | 62% (+ 22% SM) |
| 4 | BrCCl2CCl2Br | 42% (+ 11% SM & 30% 7b)c |
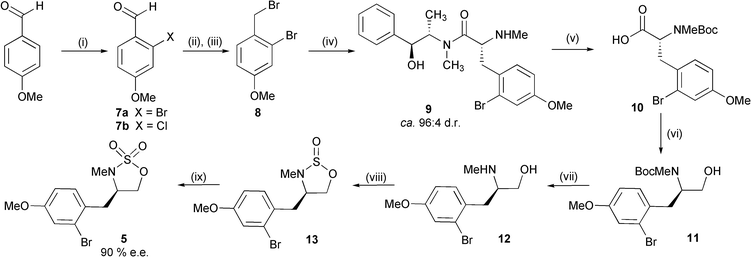 | ||
| Scheme 3 Reagents and conditions: i, n-BuLi, trimethylethylenediamine, THF, −20 °C, then n-BuLi, −20 °C, then CBr4 (62%) (see Table 1), −78 °C to r.t.; ii, NaBH4, MeOH, THF, r.t.; iii, PBr3, Et2O, 0 °C to r.t. (95% from 7a); iv, (S,S)-(+)-pseudoephedrine sarcosinamide, LDA (1.95 eq.), LiCl (6 eq.), THF, 0 °C, 1 h then bromide 8 (1.05 eq.), 0 °C, (77%); v, NaOH, MeOH, H2O, reflux, then Boc2O, NaHCO3, dioxane, 0 °C to r.t., (99%); vi, EtCO2Cl, Et3N, THF, 0 °C, then NaBH4, H2O; vii, TFA, CH2Cl2, (85% from 10); viii, SOCl2, imidazole, Et3N, CH2Cl2, 0 °C; ix, RuCl3 (0.15 mol%), NaIO4, H2O, EtOAc, 0 °C (81% from 12). | ||
Aldehyde 7a was reduced (NaBH4) and brominated (PBr3) to afford benzyl bromide 8 in 95% yield over the two steps.‡ We initially chose to employ an auxiliary controlled protocol to install the key stereocentre present in sulfamidate 5; Myers has previously outlined the use of pseudoephedrine sarcosinamide as a precursor to N-methylated amino acid derivatives.7 Accordingly, alkylation of the dianion derived from (S,S)-(+)-pseudoephedrine sarcosinamide with bromide 8 generated adduct 9 in 77% yield and in approximately 92% d.e. (as estimated by HPLC analysis of peracetylated material).§ Direct hydrolytic cleavage (H2O, dioxane, reflux) of the auxiliary was possible but we were only able to isolate the corresponding amino acid in moderate (45%) yield. In addition, attempted reduction of this species with LiAlH4 to the requisite amino alcohol 12 was unsuccessful due to concomitant reductive debromination. We therefore chose to employ Myers’ alternative base-induced auxiliary cleavage protocol and, after requisite Boc protection, were able to isolate adduct 10 in excellent yield (99%). Reduction of this substrate (to 11) was successful under mixed anhydride conditions and, following Boc deprotection, amino alcohol 12 was isolated in 85% overall yield from 10. This material was converted to cyclic sulfamidate 5via the corresponding cyclic sulfamidite 13 in good overall yield (81%). Critically, the use of EtOAc–H2O8 as solvent for the oxidation step was absolutely essential.¶ When we employed a more commonly used MeCN–H2O solvent system, yields of 5 were significantly lower (14–55%) and irreproducible, presumably due to competing oxidation of the 4-methoxybromobenzyl substituent (Table 2). It therefore appears that the chemoselectivity and/or reactivity of the oxidation step can be tailored via judicious choice of reaction solvent. At this stage we were able to determine the enantiopurity of 5 as 90% e.e. by chiral HPLC (using the corresponding racemate as a standard); this reflects upon the diastereoselectivity of the alkylation step (8 → 9). The route outlined in Scheme 3 proceeds in 9 steps and in 29% overall yield.
| Entrya | Solvent | NaIO4 eq. | Scale | Time | Yieldb |
|---|---|---|---|---|---|
| a All reactions were performed at 0 °C using 0.15 mol% RuCl3. b Isolated yield. | |||||
| 1 | 1 : 1 MeCN–H2O | 1.5 | 0.78 mmol | 2.5 hrs | 14% |
| 2 | 1 : 1 MeCN–H2O | 1.0 | 0.26 mmol | 50 min | 55% |
| 3 | 1 : 1 MeCN–H2O | 1.0 | 1.14 mmol | 30 min | 18% |
| 4 | 2 : 1 MeCN–H2O | 1.0 | 0.98 mmol | 20 min | 40% |
| 5 | 1 : 1 EtOAc–H2O | 1.0 | 0.80 mmol | 10 min | 82% |
Given the level of enantiomeric excess (90%) associated with 5 and the length of the sequence, we chose to focus upon an alternative hydrogenation strategy to synthesise the key amino alcohol 12. Condensation of bromide 7a with ethyl isocyanoacetate afforded Knoevenagel adduct 14 in 41% yield and as a mixture of geometric isomers (1 : 1 E–Z) (Scheme 4). Unfortunately we were unable to achieve reduction of this substrate (to give 15) with a variety of RhL* systems.9|| Hydrogenation of an N-Boc variant 16 was, however, successful (Scheme 5).
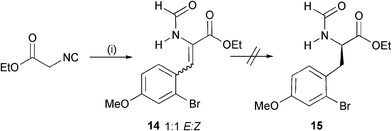 | ||
| Scheme 4 Reagents and conditions: i, 7a, NaH, THF, 0 °C to r.t. (41%). | ||
![Reagents and conditions i, BocNHCH(PO(OMe2))CO2Me, N,N,N′,N′-tetramethylguanidine (TMG), CH2Cl2 (99%); ii, [((R,R)–Et-DuPHOS)Rh(COD)]BF4 (1.5 mol%), H2 (5 bar), MeOH (100%).](/image/article/2007/OB/b614999e/b614999e-s5.gif) | ||
| Scheme 5 Reagents and conditions i, BocNHCH(PO(OMe2))CO2Me, N,N,N′,N′-tetramethylguanidine (TMG), CH2Cl2 (99%); ii, [((R,R)–Et-DuPHOS)Rh(COD)]BF4 (1.5 mol%), H2 (5 bar), MeOH (100%). | ||
Reaction of bromide 7a with Boc-α-phosphonoglycine trimethyl ester in the presence of TMG yielded Boc protected dehydroamino ester 16 in essentially quantitative yield and as a single geometric isomer (>99 : 1 Z–E) which was assigned on the basis of extensive literature precedent.10 Reduction of this species was possible and, of the catalysts evaluated (Table 3), a Rh-Et-DuPHOS system delivered the best conversion and asymmetric induction (>98% e.e. as determined by chiral HPLC) to afford amino acid derivative 17 in quantitative yield.11
| Entrya | Catalyst | e.e.b | Yieldc |
|---|---|---|---|
| a Reaction conditions: catalyst (1.5 mol%), H2 (5 bar), r.t., MeOH, 36 h. b Determined by chiral HPLC using a racemic standard prepared with Wilkinson's catalyst. c Isolated yield. | |||
| 1 | ((R,R)–Et-DuPHOS)Rh(COD)]BF4 | >98% | 100% |
| 2 | ((S,S)–i-Pr-DuPHOS)Rh(COD)]BF4 | n.d. | 8% (+ 92% 16) |
| 3 | ((R,R)–Me-BPE)Rh(COD)]BF4 | 55% | 100% |
Reduction of both the ester and N-Boc moieties of 17 directly to amino alcohol 12 with LiAlH4 was not possible, due again to concomitant debromination. We therefore examined the possibility of an N-methylation,12 Boc deprotection and chemoselective (0 °C) ester reduction sequence. Although we were able to obtain amino alcohol 12 in 77% yield over these three steps, it was evident upon conversion to the cyclic sulfamidate 5 (62% e.e.) and analysis by chiral HPLC that some epimerisation had occurred, presumably during the base-mediated N-methylation step (Scheme 6).
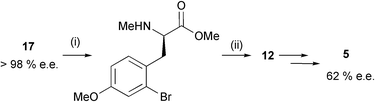 | ||
| Scheme 6 Reagents and conditions: i, NaH (1.2 eq.), MeI, DMF, r.t. then TFA (82%); ii, LiAlH4, THF, 0 °C, (93%). | ||
An alternative strategy for the conversion of amino ester 17 to the target amino alcohol 12 was devised (Scheme 7). Chemoselective reduction of ester 17 with LiAlH4 (0 °C) proceeded smoothly to afford alcohol 18 in 96% yield with the Ar–Br bond (and N-Boc group) still intact. Treatment of this species with NaH effected cyclisation to cyclic carbamate 19. Once this step was complete, as judged by TLC, methyl iodide and further NaH were added to achieve methylation to afford 20.** This intermediate was cleaved hydrolytically and the resulting amino alcohol 12 was isolated in 92% overall yield. Conversion of 12 to cyclic sulfamidate 5 proceeded smoothly using our previously developed conditions and at this stage the enantiopurity of 5 was confirmed as >98% e.e. by chiral HPLC. This sequence allowed us to synthesise the key intermediate 5 in 48% overall yield over 7 steps from p-anisaldehyde (compare against Scheme 3).
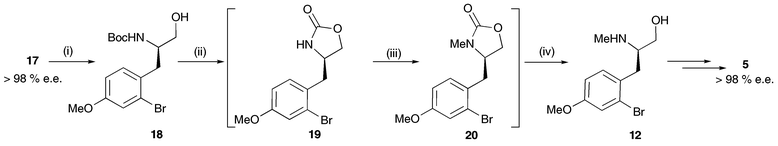 | ||
| Scheme 7 Reagents and conditions: i, LiAlH4, THF, 0 °C (96%); ii, NaH, THF; iii, NaH, MeI, THF; iv, NaOH, MeOH, reflux (92% from 18). | ||
Attention now turned to the synthesis of α-methyl lactam 4. Although we have previously reported reactions of structurally representative cyclic sulfamidates with stabilised enolates as a route to C(3)-substituted lactams, the direct introduction of α-alkyl groups, via simple alkyl ester enolates, has been unsuccessful.4b Sulfamidate 5 did, however, react efficiently with the sodium enolate of α-methyl diethyl malonate to deliver lactam 21 after hydrolysis (to cleave the intermediate N-sulfate) and lactamisation (Scheme 8). Hydrolysis and subsequent decarboxylation afforded lactam 4 as a 2 : 1 mixture of diastereomers in 65% yield from cyclic sulfamidate 5; this sequence was conducted efficiently without purification of intermediate 21. Attempted cyclisation by exposure of lactam 4 to a variety of Pd-catalysed α-arylation protocols was unsuccessful.13 At lower temperatures (<100 °C) starting material remained intact and at higher temperatures (110–140 °C) only debromination was observed under a variety of conditions, including those previously employed by Cossy to α-arylate piperidinones.14a,b We also briefly investigated the possibility of cyclisation via a benzyne intermediate by treating 4 with 2 equivalents of LiTMP but this led to decomposition.15
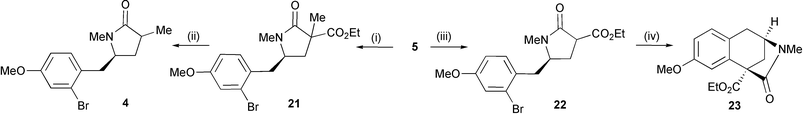 | ||
| Scheme 8 Reagents and conditions: i, methyl diethylmalonate, NaH, DMF, 40 °C then 5 M HCl; ii KOH, dioxane, reflux, then p-xylene, reflux (65% from 5); iii, diethylmalonate, NaH, DMF, r.t. then 5 M HCl (80%); iv, Pd(dba)2 (12 mol%), dppp (18 mol%), t-BuOK, PhMe, 130 °C (17%). | ||
More stabilised enolates are generally better coupling partners for Pd-catalysed α-arylation. To this end we treated cyclic sulfamidate 5 with diethyl malonate to afford α-ester lactam 22 in 80% yield and as a 2 : 1 mixture of diastereomers. This species did indeed undergo successful Pd-catalysed cyclisation at higher temperatures to afford tricycle 23 but the reaction was disappointingly inefficient.†† A screen of Pd source (Pd(OAc)2, Pd(dba)2), solvent (DMF, dioxane, p-xylene, toluene), base (Cs2CO3, t-BuONa, t-BuOK), temperature (100–150 °C) and ligand (dppf, dppp, dppe, Xantphos, BINAP) failed to rectify this problem and at best we were able to obtain the tricyclic adduct 23 in only 17% yield (Scheme 8). In all cases, the main byproducts were debrominated and decarboxylated adducts. Decarboxylation could potentially occur either via hydrolysis of ester of 22 to the corresponding acid (due to adventitious water) or via a Krapcho-type process promoted by released bromide.16‡‡
A potentially more robust approach to 1 involves Pd catalysed cyclisation via an alkene (i.e. an intramolecular reductive Heck reaction). Cyclic sulfamidate 5 thus reacted with the potassium enolate of triethylphosphonoacetate to afford, after hydrolysis and lactamisation, α-phosphono lactam 24 in 84% yield (2 : 1 d.r. at C(3)) (Scheme 9). This compound then underwent efficient Horner–Wadsworth–Emmons reaction with paraformaldehyde to afford exo-alkene 25 in 74% yield; in our hands 25 proved to be sensitive to chromatography and was used in subsequent stages without purification. Exposure of 25 to reductive Heck conditions did not lead to cyclisation but routinely resulted in debromination and/or double bond isomerisation within the starting material.17 However, the desired cyclisation could be achieved under radical conditions. Generation of the requisite aryl radical by slow addition (over 1.5 h) of a solution of Bu3SnH and AIBN in PhH to 25 under high dilution conditions resulted in cyclisation to deliver the target tricycle 26 in 62% yield. Reduction of the lactam carbonyl using conditions previously reported by Funk on a synthesis of racemic 1 yielded amine 27 ([α]20D +8.3 (c 0.5, CHCl3); lit. [α]28D +8.1 (c 1.2, CHCl3),3h [α]20D +9.4 (c 0.3, CHCl3),3j [α]20D +8.7 (c 1.06, CHCl3)3n). This constitutes a formal asymmetric synthesis of (−)-1; O-demethylation of amine 27 has been reported by a number of groups in yields ranging from 57–86%.
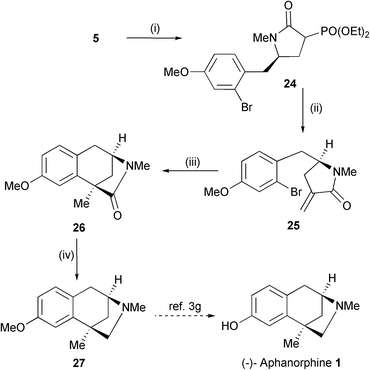 | ||
| Scheme 9 Reagents and conditions: i, (EtO)2OPCH2CO2Et, t-BuOK, THF, 40 °C then 5 M HCl (84%); ii, NaH, paraformaldehyde, THF (74%); iii, Bu3SnH–AIBN (added over 1.5 h), PhH, reflux (62% + 18% of 30); iv, LiAlH4, THF (93%). | ||
The aryl radical cyclisation (25 → 26) is noteworthy, particularly in light of previous related work by Ishibashi3j,m who used a similar strategy to access the tricyclic core of 1. In their system a thio-substituted radical acceptor, as in 28, was required to achieve an efficient cyclisation (Scheme 10). Model studies on the corresponding des-methoxy series had shown that in the absence of such an activating group, 1,5-hydrogen atom abstraction competed leading to the isolation of endo-alkene 29. We also observe the formation of a similar byproduct 30 but in our system there is no requirement for additional activation of the alkene to achieve an efficient cyclisation to form 26. Interestingly the amount of 30 does increase if the Bu3SnH is added over a shorter time period (e.g. addition over 45 minutes affords 26 in 50% yield along with 25% of 30). This observation strongly suggests that 1,5-hydrogen atom abstraction may not be the only pathway corresponding to the formation of 30 and that direct isomerisation may also contribute. We have also briefly investigated using TMS3SiH to carry out this cyclisation but this has proved to be less efficient.18
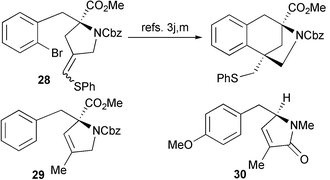 | ||
| Scheme 10 | ||
Conclusions
In summary, we have demonstrated the applicability of cyclic sulfamidate 5 as a key intermediate for the facile synthesis and evaluation of a range of cyclisation precursors leading to the tricyclic core of 1. The availability of enantioenriched alkylating agents, such as 5, was also demonstrated by applying two efficient and general literature methods for the asymmetric synthesis of amino acids. Pd-catalysed cyclisation of the three different precursors investigated was largely unsuccessful and is perhaps a reflection of the steric demands of forming the C(1)–C(9a) bond associated with 1. Aryl radical cyclisation was, however, effective and allowed the completion of an efficient asymmetric synthesis of 1 in 15% overall yield in 12 steps from a commercial starting material. This demonstration of the versatile and modular nature of this cyclic sulfamidate mediated lactam methodology should open the way to its application in other target directed projects.Experimental
General
General experimental details have been reported recently.4c,5 Experimental procedures associated with the synthesis of (−)-1 which were included as Electronic Supporting Information within ref. 5 have been omitted from here.Via Compound 10. To a solution of acid 10 (6.06 g, 16.1 mmol) in THF (37 mL) at 0 °C was added Et3N (2.47 mL, 17.7 mmol) and then ethyl chloroformate (1.70 mL, 17.7 mmol) causing the immediate formation of a colourless precipitate. The mixture was stirred at 0 °C for 50 minutes and was then filtered (washing with THF (37 mL)). The filtrate was then added dropwise to an ice-cooled (0 °C) suspension of NaBH4 (1.53 g, 40.3 mmol) in water (37 mL) causing vigorous gas evolution. The mixture was stirred at 0 °C for 2.5 h and was then poured into aq. 0.5 M HCl (120 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organic extracts were washed with brine (100 mL), dried (Na2SO4) and then concentrated in vacuo to afford 11 as a colourless oil. This residue was dissolved in CH2Cl2 (30 mL) and then TFA (30 mL) was added resulting in immediate bubbling. The mixture was stirred at r.t. for 0.5 h and then concentrated in vacuo. The residue was partitioned between CH2Cl2 (100 mL), saturated aq. NaHCO3 (100 mL) and water (50 mL). The organic portion was isolated and the aqueous portion was further extracted with CH2Cl2 (2 × 80 mL). The combined organic extracts were then dried (Na2SO4) and concentrated in vacuo to afford amino alcohol 12 (3.77 g, 85%) as a colourless wax. This material was used for the synthesis of cyclic sulfamidate 5 (90% e.e.) without further purification.5
Characterisation of 12 (in >98% e.e.) has been reported previously.5
Acknowledgements
We acknowledge EPSRC and GSK for financial support of this research programme, and Dow Pharma for providing the Rh hydrogenation catalysts used in this studyReferences
- N. Gulavita, A. Hori, Y. Shimizu, P. Laszlo and J. Clardy, Tetrahedron Lett., 1988, 29, 4381 CrossRef CAS.
- For a review of the synthesis of pentazocine 2 and related benzomorphans, see: D. C. Palmer and M. J. Strauss, Chem. Rev., 1977, 77, 1 Search PubMed.
- For total and formal syntheses of (−)-1, see: (a) S. Takano, K. Inomata, T. Sato, M. Takahashi and K. Ogasawara, J. Chem. Soc., Chem. Commun., 1990, 290 RSC; (b) A. N. Hulme, S. S. Henry and A. I. Meyers, J. Org. Chem., 1995, 60, 1265 CrossRef; (c) K. O. Hallinan and T. Honda, Tetrahedron, 1995, 51, 12211 CrossRef CAS; (d) A. Fadel and P. Arzel, Tetrahedron: Asymmetry, 1995, 6, 893 CrossRef CAS; (e) A. I. Meyers, W. Schmidt and B. Santiago, Heterocycles, 1995, 40, 525 CrossRef CAS; (f) M. Node, H. Imazato, R. Kurosaki, Y. Kawano, T. Inoue, K. Nishide and K. Fuji, Heterocycles, 1996, 42, 811 CrossRef CAS; (g) S. Shiotani, H. Okada, K. Nakamata, T. Yamamoto and F. Sekino, Heterocycles, 1996, 43, 1031 CrossRef CAS; (h) M. Shimizu, T. Kamikubo and K. Ogasawara, Heterocycles, 1997, 46, 21 CrossRef CAS; (i) A. Fadel and P. Arzel, Tetrahedron: Asymmetry, 1997, 8, 371 CrossRef CAS; (j) O. Tamura, T. Yanagimachi, T. Kobayashi and H. Ishibashi, Org. Lett., 2001, 3, 2427 CrossRef CAS; (k) K. Tanaka, T. Taniguchi and K. Ogasawara, Tetrahedron Lett., 2001, 42, 1049 CrossRef CAS; (l) A. S. ElAzab, T. Taniguchi and K. Ogasawara, Heterocycles, 2002, 56, 39 CrossRef CAS; (m) O. Tamura, T. Yanagimachi and H. Ishibashi, Tetrahedron: Asymmetry, 2003, 14, 3033 CrossRef CAS; (n) H. Zhai, S. Luo, C. Ye and Y. Ma, J. Org. Chem., 2003, 68, 8268 CrossRef CAS; (o) S. K. Taylor, M. Ivanovic, L. J. Simons and M. M. Davis, Tetrahedron: Asymmetry, 2003, 14, 743 CrossRef CAS; (p) Y. Kita, J. Futamura, Y. Ohba, Y. Sawama, J. K. Ganesh and H. Fujioka, J. Org. Chem., 2003, 68, 5917 CrossRef CAS; (q) H. Hu and H. Zhai, Synlett, 2003, 2129 CAS; (r) For syntheses of (+)-1, see: S. Takano, K. Inomata, T. Sato and K. Ogasawara, J. Chem. Soc., Chem. Commun., 1989, 1591 Search PubMed; (s) M. Katoh, H. Inoue, A. Suzuki and T. Honda, Synlett, 2005, 2820 CAS; (t) For syntheses of (±)-1, see: T. Honda, A. Yamamoto, Y. Cui and M. Tsubuki, J. Chem. Soc., Perkin Trans. 1, 1992, 531 Search PubMed; (u) J. R. Fuchs and R. L. Funk, Org. Lett., 2001, 3, 3923 CrossRef CAS.
- (a) A. J. Williams, S. Chakthong, D. Gray, R. M. Lawrence and T. Gallagher, Org. Lett., 2003, 5, 811 CrossRef CAS; (b) J. F. Bower, J. Švenda, A. J. Williams, J. P. H. Charmant, R. M. Lawrence, P. Szeto and T. Gallagher, Org. Lett., 2004, 6, 4727 CrossRef CAS; (c) J. F. Bower, S. Chakthong, J. Švenda, A. J. Williams, R. M. Lawrence, P. Szeto and T. Gallagher, Org. Biomol. Chem., 2006, 4, 1868 RSC.
- J. F. Bower, P. Szeto and T. Gallagher, Chem. Commun., 2005, 5793 RSC.
- (a) Y. Lear and T. Durst, Can. J. Chem., 1997, 75, 817 CAS; (b) D. L. Comins and J. D. Brown, J. Org. Chem., 1984, 49, 1078 CrossRef CAS.
- (a) A. G. Myers, J. L. Gleason and T. Y. Yoon, J. Am. Chem. Soc., 1995, 117, 8488 CrossRef CAS; (b) A. G. Myers, J. L. Gleason, T. Y. Yoon and D. W. Kung, J. Am. Chem. Soc., 1997, 119, 656 CrossRef CAS; (c) A. G. Myers and J. L. Gleason, Org. Synth., 1999, 76, 57 CAS.
- D. Alker, K. J. Doyle, L. M. Harwood and A. McGregor, Tetrahedron: Asymmetry, 1990, 1, 877 CrossRef CAS.
- After completion of this work, a report on the successful Rh-catalysed asymmetric hydrogenation of N-formyl dehydroamino esters, related to 14, was published, see: L. Panella, A. M. Aleixandre, G. J. Kruidhof, J. Robertus, B. L. Feringa, J. G. de Vries and A. J. Minnaard, J. Org. Chem., 2006, 71, 2026 Search PubMed.
- U. Schmidt, A. Lieberknecht and J. Wild, Synthesis, 1984, 53 CrossRef CAS.
- M. J. Burk, J. E. Feaster, W. A. Nugent and R. L. Harlow, J. Am. Chem. Soc., 1993, 115, 10125 CrossRef CAS.
- J. R. Coggins and N. L. Benoiton, Can. J. Chem., 1971, 49, 1968 CAS.
- For a review on Pd catalysed α-arylation reactions, see: D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234 Search PubMed.
- (a) J. Cossy, A. de Filippis and D. G. Pardo, Org. Lett., 2003, 5, 3037 CrossRef CAS; (b) J. Cossy, A. de Filippis and D. G. Pardo, Synlett, 2003, 2171 CrossRef CAS; (c) For a recent example of an intramolecular Pd-catalysed α-arylation reaction employing a piperidinone, see: C. Botuha, C. M. S. Galley and T. Gallagher, Org. Biomol. Chem., 2004, 2, 1825 Search PubMed.
- J. D. Stewart, S. C. Fields, K. S. Kochhar and H. W. Pinnick, J. Org. Chem., 1987, 52, 2110 CrossRef CAS.
- A. P. Krapcho, G. A. Glynn and B. J. Grenon, Tetrahedron Lett., 1967, 8, 215 CrossRef.
- For two recent applications of intramolecular reductive Heck couplings in natural product synthesis, see: (a) M. Ichikawa, M. Takahashi, S. Aoyagi and C. Kibayashi, J. Am. Chem. Soc., 2004, 126, 16553 CrossRef CAS; (b) B. M. Trost, O. R. Thiel and H. C. Tsui, J. Am. Chem. Soc., 2003, 125, 13155 CrossRef CAS . These processes both employ electron deficient alkenes as key components.
- B. Giese, B. Kopping and C. Chatgilialoglu, Tetrahedron Lett., 1989, 30, 681 CrossRef CAS.
- W. G. Earley, J. A. Dority, V. Kumar and J. P. Mallamo, Heterocycles, 1995, 41, 309 CrossRef CAS.
- A. K. Ghosh, J. K. Mukhopadhyaya and U. R. Ghatak, J. Chem. Soc., Perkin Trans. 1, 1997, 2747 RSC.
- S. C. Watson and J. F. Eastham, J. Organomet. Chem., 1967, 9, 165 CrossRef CAS.
Footnotes |
| † 1,2-Dibromotetrachloroethane was effective on a smaller scale (0.3 mmol) and only traces of chlorinated adduct 7b were observed (<10%). On a larger scale, chlorination is most likely promoted by inferior reaction exotherm control during addition of the brominating agent. |
| ‡ Bromination is best conducted using reagent grade Et2O; anhydrous Et2O slows the reaction and results in slightly diminished yields of 8 (85% vs. 95%). |
| § We have been unable to enrich this d.e. by chromatography or crystallisation. |
| ¶ The oxidation step is most efficient on a small scale (100 mg; 82%) but can be conducted on larger scales (2 g) although yields were more variable (57–74%). |
| || The reason for the failure of this reaction is at present unclear.9 |
| ** The intermediacy of 19 and 20 is inferred although analogous intermediates have been isolated on a model des-bromo series. |
| †† At lower temperature (<100 °C) either oxidative addition of Pd(0) into the Ar–Br bond is slow or cyclisation does not occur. |
| ‡‡ An attempt was made to sequester released bromide by addition of AgOTf but this led to decomposition. |
| This journal is © The Royal Society of Chemistry 2007 |