Ti-direct, powerful, stereoselective aldol-type additions of esters and thioesters to carbonyl compounds: application to the synthesis and evaluation of lactone analogs of jasmone perfumes†
Ryohei
Nagase
,
Noriaki
Matsumoto
,
Kohei
Hosomi
,
Takahiro
Higashi
,
Syunsuke
Funakoshi
,
Tomonori
Misaki
and
Yoo
Tanabe
*
Department of Chemistry, School of Science and Technology, Kwansei Gakuin University, 2-1 Gakuen, Sanda, Hyogo 669-1337, Japan. E-mail: tanabe@kwansei.ac.jp; Fax: +81-79-565-9077; Tel: +81-79-565-8394
First published on 20th November 2006
Abstract
An efficient TiCl4–Et3N or Bu3N-promoted aldol-type addition of phenyl and thiophenyl esters or thioaryl esters with aldehydes and ketones was performed (total 46 examples). The present method is advantageous from atom-economical and cost-effective viewpoints; good to excellent yields, moderate to good syn-selectivity, substrate variations, reagent availability, and simple procedures. Utilizing the present reaction as the key step, an efficient short synthesis of three lactone [2(5H)-furanone] analogs of jasmine perfumes was performed. Among them, the lactone analog of cis-jasmone had a unique perfume property (tabac).
Introduction
The aldol-type addition of simple esters with aldehydes and ketones (carbonyl acceptor) has attained an important position in organic syntheses due to its broad utility, that is, a straightforward method to access useful β-hydroxy esters.1 In general, this reaction is categorized into two types: (i) the direct method of metal enolates generated by strong basic reagents [e.g., LDA and MHMDS (M = Li, Na, K) (metal exchanged enolates are also employable)] to react with the carbonyl acceptor, and (ii) the indirect method of ketene silyl acetals derived from carboxylic esters to react with the carbonyl acceptor promoted by Lewis acids or other catalysts (the Mukaiyama protocol).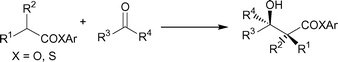
Direct aldol-type addition of simple esters with carbonyl acceptors using Lewis acids combined with tertiary amines possesses several advantages because of its simple and atom-economical procedure, high regio- and stereoselectivity, and tolerance to basic labile functionalities. Due to the lower enolization ability of esters (pKa ∼ 25) than that of ketones or aldehydes (pKa ∼ 19–21), however, there have been only three successful methods to this objective; the use of diazabromoborane (Corey's group),2 dicyclohexyliodoborane (Brown's group),3 and boron triflate (Masamune and Abiko's group).4 From the recently recognized standpoint of process chemistry, readily available, atom-economical, and cost-effective synthetic reagents are increasingly required for the production of fine chemicals. Consistent with our continued studies on Ti-Claisen condensation5 and relevant Ti-direct aldol-type additions,6 we report herein the full details of direct, powerful, and stereoselective aldol-type additions of esters and thioesters to aldehydes and ketones promoted by a TiCl4-amine reagent.7 This paper also describes an application for the short and efficient synthesis of three lactone analogs for dihydrojasmone (known), cis- and trans-jasmone (novel), with an evaluation of their perfume property.
Results and discussion
The salient features of these Ti-mediated reactions are as follows. (i) High reaction velocities and yields. (ii) Higher atom-economy and lower cost than the indirect methods using enol silyl ethers and ketene silyl acetals. (iii) Use of readily available and low toxic metal reagents (e.g., TiCl4, ZrCl4), and use of practical amines (Et3N, Bu3N) and solvents (toluene, CH2Cl2). (iv) Toleration against basic labile functionalities. (v) Enhanced reactivity using catalytic TMSCl in rare cases.(A) Basic investigation of Ti-direct aldol-type addition of methyl propanoate to aldehydes
The initial attempt was guided by the reaction between methyl propanoate (CH3CH2CO2CH3) and PhCHO or t-BuCHO utilizing a TiCl4–Bu3N reagent (Scheme 1). The desired β-hydroxy methyl esters 1a, 1b were obtained in 75% and 82% yield, respectively. This result is in clear contrast to the method using a TiCl2(OTf)2–Et3N reagent, which resists the aldol-type addition and promotes a competitive Ti-Claisen condensation of CH3CH2CO2CH3, supported by careful cross-over experiments.5a Unfortunately, the carbonyl acceptor was limited to aldehydes lacking α-protons, that is, an undesirable side self-aldol addition predominated between two aldehydes such as i-PrCHO, which has α-protons.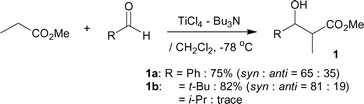 | ||
| Scheme 1 Ti-direct aldol-type addition of methyl propanoate to aldehydes. | ||
To overcome the problem, we planned to use slightly acidic phenyl esters 2, because of the higher acidity of phenyl esters than that of alkyl esters.8 Phenyl esters are readily prepared by several methods and are easily hydrolyzed under milder conditions compared with alkyl esters.9
(B) Ti-direct aldol-type addition of phenyl ester to aldehydes and ketones
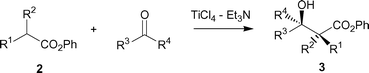
The aldol-type reaction of phenyl esters 2 with various aldehydes or ketones utilizing TiCl4–Et3N was examined. Table 1 lists the successful results. The salient features are as follows. (i) Because of the specific character of titanium enolate,10 the present method is powerful enough to conduct the addition not only to aldehydes but also to less reactive ketones, giving the desired β-hydroxy esters 3. (ii) Et3N is somewhat superior to Bu3N with regard to yield (entries 1–4) in contrast to the Ti-crossed aldol additions6b between two different ketones.11 (iii) Moderate to good syn-selectivity was obtained. (iv) This method is applicable to the reaction of less reactive phenyl 2-methylpropanoate under practical temperatures (0–5 °C) (entries 11–16).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | Product | Yield (%) | Syn–antib |
| a Carried out in CH2Cl2 at −78 °C (entries 1–10) and at 0–5 °C (entries 11–16). Molar ratio 2–ketone or aldehyde–TiCl4–Et3N = 1 : 1.2 : 1.2 : 1.4 (entries 1–10) and 2–ketone or aldehyde–TiCl4–Et3N = 1 : 1.2 : 1.5 : 2.0 (entries 11–16). b Determined by 1H NMR of the crude product. c Use of Bu3N instead of Et3N. d Yield based on its TMS ether. | |||||||
| 1 | Me | H | Ph | H | 3a | 80 | 82 : 18 |
| 2c | 3a | 75 | 85 : 15 | ||||
| 3 | Me | H | n-Pr | H | 3b | 84 | 84 : 16 |
| 4c | 3b | 76 | 84 : 16 | ||||
| 5 | Me | H | i-Pr | H | 3c | 79 | 89 : 11 |
| 6 | Me | H | Ph | Et | 3d | 83 | 64 : 36 |
| 7 | Me | H | Et | Et | 3e | 77 | — |
| 8 | Me | H | Ph | CH2Cl | 3f | 86d | 68 : 32 |
| 9 | Bu | H | Ph | H | 3g | 76 | 84 : 16 |
| 10 | Bu | H | Ph | Et | 3h | 87 | 65 : 35 |
| 11 | Me | Me | Ph | H | 3i | 87 | — |
| 12 | Me | Me | n-Pr | H | 3j | 80 | — |
| 13 | Me | Me | i-Pr | H | 3k | 80 | — |
| 14 | Me | Me | Ph | Et | 3l | 81 | — |
| 15 | Me | Me | Et | Et | 3m | 73 | — |
| 16 | Me | Me | Ph | CH2Cl | 3n | 79 | — |
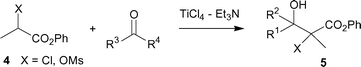
Encouraged by this result, we next investigated the aldol-type addition of readily available phenyl 2-chloro and 2-mesyloxypropanoates (4; CH3XCHCO2Ph, X = Cl, OMs) with aldehydes or ketones. Table 2 lists the successful results. α-Halogenated esters are labile under basic conditions and generally undergo further Darzens-type reactions to form α,β-epoxyesters.12
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X | R1 | R2 | Product | Yield (%) | Drb,c |
| a Carried out in CH2Cl2 at 0–5 °C (entries 1–4) and at −78 °C (entries 5, 6). Molar ratio 4–ketone or aldehyde–TiCl4–Et3N = 1 : 1.2 : 1.2 : 1.4. b Determined by 1H NMR of the crude product. c Syn/anti was not assigned. | ||||||
| 1 | Cl | Ph | H | 5a | 88 | (70 : 30) |
| 2 | Cl | i-Pr | H | 5b | 86 | (74 : 26) |
| 3 | Cl | Et | Et | 5c | 69 | — |
| 4 | Cl | Ph | Et | 5d | 75 | (74 : 26) |
| 5 | OMs | Ph | H | 5e | 89 | (89 : 11) |
| 6 | OMs | Et | Et | 5f | 60 | — |
Note that the reaction using CH3ClCHCO2CH3 instead of the corresponding phenyl esters 4 resulted in oxidative self-coupling addition at the α-carbon to give dimeric product 6 (Scheme 2), as described by the Kise, Matsumura, and Periasamy groups.13 Accordingly, from the synthetic view, especially for aldol chemistry, the use of phenyl esters has an advantage over that of methyl esters.
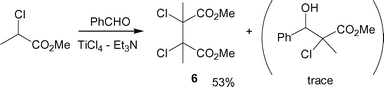 | ||
| Scheme 2 Ti-promoted oxidative coupling of methyl 2-chloropropanoate. | ||
(C) Ti-direct aldol-type addition of phenylthio esters to aldehydes and ketones
The use of phenylthio esters 7 instead of phenyl esters 2 has a notable advantage, because of inherently feasible enolate formation.14Table 3 lists the successful results. As anticipated, Ti-enolates of 7 were smoothly generated because of the higher acidity of the α-proton of 7 than that of 2. The salient features are as follows. (i) With the exception of only a few cases, these reactions proceeded smoothly in excellent yields to afford the desired β-hydroxyl thioesters 8. (ii) In contrast to the case using phenyl esters 2, Bu3N was slightly superior to Et3N with regard to yield (entries 1, 2). (iii) α,α-Disubstituted phenylthio esters successfully underwent the reaction at 0–5 °C (entries 9–12).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | Product | Yield (%) | Syn–antib |
| a Carried out in CH2Cl2 at −78 °C (entries 1–8) and at 0–5 °C (entries 9–12). Molar ratio 7–aldehyde (ketone)–TiCl4–Bu3N = 1 : 1.2 (1.5) : 1.2 : 1.4. b Determined by 1H NMR of the crude product. c Use of Et3N instead of Bu3N. d Syn/anti was not assigned. | |||||||
| 1 | Me | H | Ph | H | 8a | 99 | 86 : 14 |
| 2c | 8a | 94 | 86 : 14 | ||||
| 3 | Me | H | n-Pr | H | 8b | 98 | 82 : 18 |
| 4 | Me | H | i-Pr | H | 8c | 96 | 81 : 19 |
| 5 | Me | H | CH2CH2Ph | H | 8d | 99 | 83 : 17 |
| 6 | Me | H | Et | Et | 8e | 77 | — |
| 6 | Me | H | Ph | Et | 8f | 98 | 77 : 23 |
| 7 | Bu | H | Ph | H | 8g | 98 | 77 : 23 |
| 8 | Bu | H | Et | Et | 8h | 78 | — |
| 9 | Me | Me | Ph | H | 8i | 80 | — |
| 10 | Me | Me | n-Pr | H | 8j | 76 | — |
| 11 | Me | Cl | Ph | H | 8k | 97 | (70 : 30)d |
| 12 | Me | Cl | n-Pr | H | 8l | 86 | (80 : 20)d |
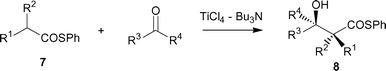
The substituent effect was investigated to improve the yield and stereoselectivity. Table 4 lists the results. Among the aryl thioesters screened, 4-nitrophenyl propanethioate 9 produced better results than phenylthio ester 8a with PhCHO regarding the syn-selectivity (entry 7). The use of n-PrCHO, i-PrCHO, and PhCOEt as electrophiles, however, did not notably improve the results (entries 9–12).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ar | R1 | R2 | Product | Yield (%) | Syn–antib |
| a Carried out in CH2Cl2 at −78 °C. Molar ratio 9–aldehyde (ketone)–TiCl4–Bu3N = 1 : 1.2 (1.5) : 1.2 : 1.4. b Determined by 1H NMR of the crude product. | ||||||
| 1 | Ph | Ph | H | 8a | 99 | 86 : 14 |
| 2 | (4-OMe)Ph | 10a | 95 | 80 : 20 | ||
| 3 | (4-t-Bu)Ph | 10b | 99 | 82 : 18 | ||
| 4 | (2-Cl)Ph | 10c | 92 | 90 : 10 | ||
| 5 | (3-Cl)Ph | 10d | 95 | 86 : 14 | ||
| 6 | (4-Cl)Ph | 10e | 96 | 86 : 14 | ||
| 7 | (4-NO2)Ph | 10f | 99 | 94 : 6 | ||
| 8 | n-Pr | H | 10g | 97 | 74 : 26 | |
| 9 | i-Pr | H | 10h | 92 | 67 : 33 | |
| 10 | Ph | Et | 10i | 90 | 79 : 21 | |
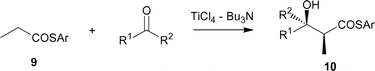
(D) Synthesis of lactone [2(5H)-furanone] analogs for three jasmone perfumes
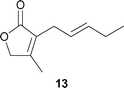
The present Ti-direct aldol-type addition was successfully applied to the synthesis of three lactone [2(5H)-furanone] analogs 11–13 of jasmone, which is a typical jasmine perfume (Fig. 1). Lactone analog 12 of cis-jasmone is a particularly promising candidate for the synthetic isoster. The Givordan group reported the synthesis of lactone analog 11 of dihydrojasmone utilizing the Reformatsky reaction as the key step.15 This method, however, is difficult to apply for the synthesis of 12 bearing a double bond in R. Our interest in the practical short synthesis of useful natural perfumes such as cis-jasmone and (R)-muscone,5h (Z)-civetone,5d,f (R)-mintlactone and (R)-menthofuran,6c utilizing Ti-mediated C–C bond forming reactions led us to investigate the general synthesis for lactone analogs 11–13.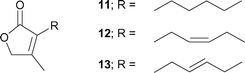
The synthesis of dihydrojasmone lactone analog 11 is shown in Scheme 3. Ti-direct aldol-type addition of phenyl heptanoate with commercially available 1-acetoxy-2-propanone proceeded smoothly to give aldol adduct 14 in 72% yield. The final step was performed through a convenient one-pot deprotection and intramolecular cyclization with dehydration to give 11 in 70% yield. The total yield was increased compared with the reported method15 utilizing the Reformatsky reaction (34–40% → 50%).
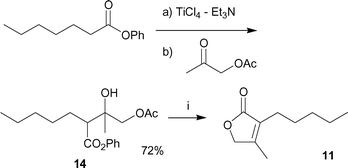 | ||
| Scheme 3 Synthesis of dihydrojasmone lactone analog 11. Reagents and conditions: (i) 1.0 M aq KOH, MeOH–THF = 2 : 1, then 1.0 M HCl, 20–25 °C (70%). | ||
Scheme 4 shows the synthesis of 12 and 13. Readily available cis- and trans-2-pentenols (Aoba alcohol analog) 15 and 16 were converted to mesylates 17 and 18, respectively, using either a conventional method (CH3SO2Cl–Et3N) or an alternative safe and practical sulfonylation for allylic alcohols (CH3SO2Cl–Et3N–Me3N·HCl).16 Alkylation using dimethyl malonate with 17 and 18 gave 19 and 20, which were subjected to hydrolysis and decarboxylation to give acids 21 and 22, respectively. Acids 21 and 22 were converted to phenyl esters 23 and 24, by conventional esterification.17 The key direct Ti-aldol-type addition of 23 and 24 with AcOCH2COCH3 gave adducts 25 (83%) and 26 (70%), respectively. In a similar one-pot procedure, novel cis- and trans-lactone analogs 12 (67%) and 13 (65%) was successfully synthesized.
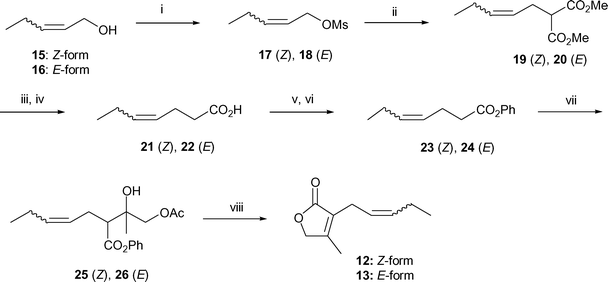 | ||
| Scheme 4 Synthesis of cis- and trans-jasmone lactone analogs 12 and 13. Reagents and conditions: (i) MsCl, Et3N, Et2O, 0–5 °C or MsCl, Et3N–cat. Me3N·HCl, toluene, 0–5 °C. (ii) Dimethyl malonate, NaH, DMF, 20–25 °C. (iii) 5 M KOH aq, MeOH–THF = 2 : 1, reflux. (iv) Heat at ca. 150 °C. (v) SOCl2, cat. DMF, hexane, reflux. (vi) PhOH, Et3N, MeCN, 0–5 °C. (vii) TiCl4, Et3N, CH2Cl2, then AcOCH2COCH3, −78 °C. (viii) 1.0 M aq KOH, MeOH–THF = 2 : 1, then 1.0 M aq HCl, 20–25 °C. | ||
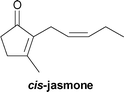
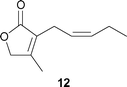
The perfume properties of the lactone analogs 11, 12, and 13 were evaluated by the Takasago International Corporation. The results are listed in Table 5. Note that the perfume of 12 showed a unique property for men's fragrance (tabac).
| Compounds | Odour description |
|---|---|
|
Floral, green, jasmine, spicy, fruity |

|
Floral, green, fruity, fatty, lactonic |
|
Floral, green, tuberose, fruity, tabac, lactonic |
|
Floral, green, fruity, jasmine, lactonic |
In conclusion, we achieved a simple and efficient Ti-direct aldol-type addition of phenyl and thiophenyl esters with aldehydes and ketones. Application of the present method for synthesizing three lactone analogs 11, 12, and 13 was performed. Evaluation of these analogs revealed that cis-jasmone analog 12 has a notable perfume property. The present method will provide a new avenue for the aldol-type reaction using esters to obtain a variety of β-hydroxy esters and lactones [2(5H)-furanones].
Experimental
Melting points were determined on a hot stage microscope apparatus (Yanagimoto) and were uncorrected. NMR spectra were recorded on a JEOL DELTA 300 spectrometer, operating at 300 MHz for 1H NMR and 75 MHz for 13C NMR. Chemical shifts (δ ppm) in CDCl3 were reported downfield from TMS (=0) for 1H NMR. For 13C NMR, chemical shifts were reported on a scale relative to CDCl3 (77.00 ppm) as an internal reference. IR spectra were recorded on a JASCO FT/IR-5300 spectrophotometer.Data of known and new compounds 1a,18 1b,19 3a,20 3b,20 3c,20 3h, 3i,22 3k, 3m, 5d, 8a,23 8b,24 8c,23 8d,23 8f, 8h, 8i,25 8l, 10a, 10b, 10c, 10d, 10e, 10f, 10g, 10h, 10i, 11,15 20,25 22, 24, 19, 2125 and 23 are described in the electronic supporting information.†
Ti-direct aldol-type addition of methyl propanoate to PhCHO or t-BuCHO (Scheme 1)
Bu3N (475 µL, 2.0 mmol) and TiCl4 (165 µL, 1.5 mmol) were successively added to a stirred solution of methyl propanoate (88 mg, 1.0 mmol) and an aldehyde (PhCHO; 127 mg or t-BuCHO; 103 mg, 1.2 mmol) in CH2Cl2 (2.0 mL) at 0–5 °C under an Ar atmosphere, followed by being stirred at the same temp. for 2 h. The mixture was poured into ice water, which was extracted twice with Et2O. The combined organic phase was washed with water, brine, dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2-column chromatography to give the desired β-hydroxy ester 1a (146 mg, 75%, syn–anti = 65 : 35) or 1b (143 mg, 82%, syn–anti = 81 : 19).General procedure of Ti-direct aldol-type addition of α-monoalkylated phenyl esters to aldehydes or ketones (Table 1, entries 1–10)
TiCl4 (132 µL, 1.2 mmol) and Et3N (142 mg, 1.4 mmol) in CH2Cl2 (0.5 mL) were successively added to a stirred solution of a phenyl ester (1.0 mmol) in CH2Cl2 (1.5 mL) at −78 °C under an Ar atmosphere. After stirring at the same temp. for 30 min, an aldehyde or a ketone (1.2 mmol) was added to the mixture, followed by being stirred at the same temp. for 2 h. The mixture was poured into ice water (reverse quench), which was extracted twice with Et2O. The combined organic phase was washed with water, brine, dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2-column chromatography to give desired β-hydroxy phenyl esters 3a–3h.anti-Isomer; colorless crystals; mp 74–75 °C; 1H NMR (300 MHz, CDCl3) δ 0.70 (3H, t, J = 7.2 Hz), 1.51 (3H, d, J = 7.2 Hz), 1.73 (1H, dq, J = 7.2 Hz, Jgem = 13.8 Hz), 2.02 (1H, dq, J = 7.2 Hz, Jgem = 13.8 Hz), 3.32 (1H, q, J = 7.2 Hz), 3.74 (1H, br s), 6.42–6.53 (2H, m), 7.07–7.16 (1H, m), 7.18–7.32 (3H, m), 7.33–7.42 (2H, m), 7.45–7.53 (2H, m); 13C NMR (75 MHz, CDCl3) δ 7.53, 11.99, 31.59, 47.92, 77.19, 121.17, 125.72, 125.99, 126.89, 128.09, 129.28, 145.00, 149.85, 175.74; IR (KBr) 3532, 2973, 1725, 1352, 1192, 1154, 1098, 959, 764, 704 cm−1. Anal. Calcd for C18H20O3: C, 76.03; H, 7.09, found: C, 75.8; H, 6.8%.
syn- and anti-Mixture; colorless oil; 1H NMR (300 MHz, CDCl3) δ 0.18 (anti, 9H × 1/3, s), 0.22 (syn, 9H × 2/3, s), 1.13 (syn, 3H × 2/3, d, J = 7.2 Hz), 1.18 (anti, 3H × 1/3, d, J = 7.2 Hz), 3.17 (syn, 1H × 2/3, q, J = 7.2 Hz), 3.30 (anti, 1H × 1/3, q, J = 7.2 Hz), 4.10 (syn, 1H × 2/3, d, Jgem = 12.0 Hz), 4.13 (anti, 1H × 1/3, d, Jgem = 12.0 Hz), 4.26 (syn, 1H × 2/3, d, Jgem = 12.0 Hz), 4.35 (anti, 1H × 1/3, d, Jgem = 12.0 Hz), 6.80–6.96 (2H, m), 7.09–7.53 (8H, m); 13C NMR (75 MHz, CDCl3) δ 2.33, 12.37, 12.87, 49.92, 50.56, 50.61, 50.92, 81.19, 81.40, 121.34, 125.70, 125.80, 126.16, 126.66, 127.44, 127.65, 127.86, 127.96, 129.28, 129.37, 141.32, 142.15, 150.43, 150.46, 171.77, 171.86; IR (neat) 2957, 1759, 1493, 1252, 1192, 1161, 1084, 845 cm−1. Anal. Calcd for C17H17ClO3: C, 67.00; H, 5.62, found: C, 66.7; H, 5.5%.
General procedure of Ti-direct aldol-type addition of α,α-dimethylated phenyl esters to aldehydes or ketones (Table 1, entries 11–16)
TiCl4 (165 µL, 1.5 mmol) was added to a stirred solution of α,α-disubstituted phenyl esters (1.0 mmol) and Et3N (202 mg, 2.0 mmol) in CH2Cl2 (1.5 mL) at 0–5 °C under an Ar atmosphere. After stirring at the same temp. for 30 min, an aldehyde or ketone (1.2 mmol) was added to the mixture, followed by being stirred at 0–5 °C for 2 h. The mixture was poured into ice water, which was extracted twice with Et2O. The combined organic phase was washed with water, brine, dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2-column chromatography to give desired β-hydroxy phenyl esters 3i–3n.General procedure of Ti-direct aldol-type addition of α-chloro or α-mesyloxy phenyl esters to aldehydes or ketones (Table 2)
TiCl4 (132 µL, 1.2 mmol) and Et3N (142 mg, 1.4 mmol) in CH2Cl2 (0.5 mL) were successively added to a stirred solution of α-chloro or α-mesyloxy phenyl esters (1.0 mmol) in CH2Cl2 (1.5 mL) at 0–5 °C (entries 1–4) or −78 °C (entries 5 and 6) under an Ar atmosphere. After stirring at the same temp. for 30 min, an aldehyde or ketone (1.2 mmol) was added to the mixture, followed by being stirred at the same temp. for 2 h. The mixture was poured into ice water, which was extracted twice with Et2O. The combined organic phase was washed with water, brine, dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2-column chromatography to give desired β-hydroxy phenyl esters 5a–5f.Minor product; colorless crystals; mp 40–41 °C; 1H NMR (300 MHz, CDCl3) δ 1.05 (3H d, J = 6.9 Hz), 1.10 (3H, d, J = 6.9 Hz), 1.87 (3H, s), 1.89–2.01 (1H, m), 2.43 (1H br s), 4.02 (1H, d, J = 5.9 Hz), 7.09–7.16 (2H, m), 7.23–7.31 (1H, m), 7.37–7.46 (2H, m); 13C NMR (75 MHz, CDCl3) δ 18.62, 20.88, 21.97, 30.76, 74.13, 79.43, 120.98, 126.33, 129.56, 150.44, 169.32; IR (KBr) 3541, 2971, 1745, 1487, 1250, 1194, 1161, 1098, 1046, 752 cm−1. Anal. Calcd for C13H17ClO3: C, 60.82; H, 6.67, found: C, 60.6; H, 6.64%.
Diastereomixture; colorless oil; 1H NMR (300 MHz, CDCl3) δ 1.99 (3H × 1/2, s), 2.07 (3H × 1/2, s), 3.81 (3H × 1/2, s), 3.83 (3H × 1/2, s); 13C NMR (75 MHz, CDCl3) δ 26.11, 26.61, 53.49, 53.57, 73.31, 73.44, 169.03, 169.12; IR (neat) 3005, 2957, 1744, 1449, 1383, 1258, 1094, 976 cm−1.
General procedure of Ti-direct aldol-type addition of phenylthio esters to aldehydes or ketones (Table 3)
TiCl4 (132 µL, 1.2 mmol) and Bu3N (259 mg, 1.4 mmol) in CH2Cl2 (0.5 mL) were successively added to a stirred solution of phenylthio esters (1.0 mmol) in CH2Cl2 (1.5 mL) at −78 °C (entries 1–8) or 0–5 °C (entries 9–12) under an Ar atmosphere. After stirring at the same temp. for 30 min, an aldehyde (1.2 mmol) or ketone (1.5 mmol) was added to the mixture, followed by being stirred at the same temp. for 2 h. The mixture was poured into ice water, which was extracted twice with Et2O. The combined organic phase was washed with water, brine, dried (Na2SO4) and concentrated. The obtained crude product was purified by SiO2-column chromatography to give desired β-hydroxy phenylthio esters 8a–8l.General procedure of Ti-direct aldol-type addition of arylthio esters with aldehydes or ketones (Table 4)
TiCl4 (132 mL, 1.2 mmol) and Bu3N (259 mg, 1.4 mmol) in CH2Cl2 (0.5 mL) were successively added to a stirred solution of arylthio esters (1.0 mmol) in CH2Cl2 (1.5 mL) at −78 °C under an Ar atmosphere. After stirring at the same temp. for 30 min, an aldehyde (1.2 mmol) or ketone (1.5 mmol) was added to the mixture, followed by being stirred at the same temp. for 2 h. The mixture was poured into ice water, which was extracted twice with Et2O. The combined organic phase was washed with water, brine, dried (Na2SO4) and concentrated. The obtained crude product was purified by Si2O-column chromatography to give desired β-hydroxy arylthio esters 10a–10i.Diastereomixture; colorless oil; 1H NMR (300 MHz, CDCl3) δ 0.84–0.97 (3H, m), 1.25–1.54 (6H, m), 1.33 (3H × 7/10, s), 1.37 (3H × 3/10, s), 1.57–1.79 (1H, m), 1.80–1.96 (1H, m), 2.10 (3H × 7/10, s), 2.12 (3H × 3/10, s), 2.38 (1H, br s), 2.82 (1H × 7/10, dd, J = 3.4, 11.7 Hz), 2.83 (1H × 3/10, dd, J = 3.4, 11.7 Hz), 4.09 (1H × 7/10, d, J = 11.4 Hz), 4.10 (1H × 3/10, d, J = 11.4 Hz), 4.13 (1H × 7/10, d, J = 11.4 Hz), 4.20 (1H × 3/10, d, J = 11.4 Hz), 7.05–7.12 (2H, m), 7.21–7.29 (1H, m), 7.35–7.43 (2H, m); 13C NMR (75 MHz, CDCl3) δ 13.96, 20.84, 21.49, 22.43, 23.25, 27.00, 27.59, 27.67, 31.57, 51.66, 52.62, 68.95, 70.00, 72.39, 72.64, 121.44, 121.48, 126.08, 126.12, 129.49, 150.27, 150.33, 170.81, 173.68; IR (neat) 3495, 2957, 1748, 1493, 1373, 1233, 1196, 1163, 1113, 1044 cm−1. Anal. Calcd for C18H26O5: C, 67.06; H, 8.13, found: C, 66.8; H, 8.0%.
Diastereomixture; yellow oil; 1H NMR (300 MHz, CDCl3) δ 0.97 (3H, t, J = 7.6 Hz), 1.35 (3H × 7/10, s), 1.40 (3H × 3/10, s), 2.03–2.15 (2H, m), 2.09 (3H × 7/10, s), 2.12 (3H × 3/10, s), 2.34–2.52 (1H, m), 2.63–2.77 (1H, m), 2.82 (1H, br s), 2.87 (1H × 7/10, dd, J = 4.1, 11.0 Hz), 2.89 (1H × 3/10, dd, J = 4.1, 11.4 Hz), 4.09 (1H × 7/10, d, Jgem = 11.4 Hz), 4.11 (1H × 3/10, d, Jgem = 11.4 Hz), 4.15 (1H × 7/10, d, Jgem = 11.4 Hz), 4.20 (1H × 3/10, d, Jgem = 11.4 Hz), 5.34–5.47 (1H, m), 5.50–5.62 (1H, m), 7.01–7.10 (2H, m), 7.19–7.28 (1H, m), 7.33–7.42 (2H, m); 13C NMR (75 MHz, CDCl3) δ 14.11, 20.55, 20.84, 21.74, 23.27, 24.98, 25.47, 51.51, 52.39, 68.89, 70.04, 72.26, 72.41, 121.51, 124.59, 124.82, 126.08, 129.43, 134.69, 134.78, 150.31, 150.35, 170.73, 173.37; IR (neat) 3491, 2967, 1748, 1493, 1373, 1236, 1194, 1163, 1119, 1046 cm−1. Anal. Calcd for C18H24O5: C, 67.48; H, 7.55, found: C, 67.2; H, 7.5%.
Pale yellow oil; 1H NMR (300 MHz,CDCl3) δ 1.00 (3H, t, J = 7.6 Hz), 2.04 (3H, s), 2.16 (2H, dq, J = 6.9, 7.6 Hz), 3.03 (2H, d, J = 7.2 Hz), 4.61 (2H, d, J = 1.0 Hz), 5.28–5.39 (1H, m), 5.41–5.52 (1H, m); 13C NMR (75 MHz, CDCl3) δ 12.24, 14.03, 20.54, 21.53, 72.43, 123.58, 126.08, 133.41, 156.54, 174.71; IR (neat) 2961, 1740, 1437, 1341, 1231, 1155, 1028, 970 cm−1. Anal. Calcd for C10H14O2: C, 72.26; H, 8.49; O, 19.25, found: C, 72.1; H, 8.3%.
Diastereomixture; pale yellow oil; 1H NMR (300 MHz, CDCl3) δ 0.99 (3H, t, J = 7.2 Hz), 1.34 (3H × 7/10, s), 1.38 (3H × 3/10, s), 2.05 (2H, quint., J = 7.2 Hz), 2.10 (3H × 7/10, s) 2.13 (3H × 3/10, s), 2.37–2.61 (2H, m), 2.88 (1H × 7/10, dd, J = 4.5, 11.01 Hz), 2.90 (1H × 3/10, dd, J = 4.8, 9.6 Hz), 3.11 (1H, br s), 4.08 (1H × 7/10, d, Jgem = 11.4 Hz), 4.10 (1H × 3/10, d, Jgem = 11.4 Hz), 4.14 (1H × 7/10, d, Jgem = 11.4 Hz), 4.19 (1H × 3/10, Jgem = 11.4 Hz), 5.39–5.53 (1H, m), 5.65 (1H, dt, J = 6.2, 15.5 Hz), 6.99–7.11 (2H, m), 7.19–7.28 (1H, m), 7.32–7.43 (2H, m); 13C NMR (75 MHz, CDCl3) δ 13.57, 20.84, 21.74, 23.14, 25.53, 30.32, 30.86, 51.74, 52.62, 68.95, 70.02, 72.20, 72.35, 121.44, 121.48, 124.80, 125.07, 126.07, 129.43, 135.32, 135.45, 150.29, 170.73, 173.20; IR (neat) 3497, 2965, 1734, 1493, 1373, 1238, 1194, 1163, 1047, 970 cm−1.
Yellow oil; 1H NMR (300 MHz, CDCl3) δ 0.96 (3H, t, J = 7.2 Hz), 1.94–2.06 (2H, m), 2.03 (3H, s), 2.97 (2H, d, J = 6.2 Hz), 4.62 (2H, d, J = 1.0 Hz), 5.41 (1H, dtt, J = 1.4, 6.2, 15.1 Hz), 5.55 (1H, dtt, J = 1.4, 6.2, 15.1 Hz); 13C NMR (75 MHz, CDCl3) δ 12.14, 13.46, 25.26, 26.39, 72.37, 123.60, 125.68, 133.96, 157.04, 174.69; IR (neat) 3418, 2973, 1740, 1676, 1443, 1389, 1343, 1188, 1101, 1038 cm−1.
Acknowledgements
This research was partially supported by a Grant-in-Aid for Scientific Research on Basic Areas (B) “18350056”, Priority Areas (A) “17035087” and “18037068”, and Exploratory Research “17655045” from MEXT. We appreciate the Takasago International Corporation for the evaluation of the lactone analogs.References
- (a) C. H. Heathcock, Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 2, p. 181 Search PubMed; (b) M. B. Smith and J. March, March's Advanced Organic Chemistry, Wiley, New York, 5th edn, 2001, p. 1223 Search PubMed; (c) J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press, New York, 2001, p. 689 Search PubMed.
- (a) E. J. Corey, R. Imwinkelreid, S. Pikul and Y. B. Xiang, J. Am. Chem. Soc., 1989, 111, 5493 CrossRef CAS; (b) E. J. Corey and S. S. Kim, J. Am. Chem. Soc., 1990, 112, 4976 CrossRef CAS; (c) E. J. Corey and D.-H. Lee, Tetrahedron Lett., 1993, 34, 1737 CrossRef CAS.
- H. C. Brown and K. Ganesan, Tetrahedron Lett., 1992, 33, 3421 CrossRef CAS.
- A. Abiko, J.-F. Liu and S. Masamune, J. Org. Chem., 1996, 61, 2590 CrossRef.
- (a) Y. Tanabe, Bull. Chem. Soc. Jpn., 1989, 62, 1917 CAS; (b) Y. Yoshida, R. Hayashi, H. Sumihara and Y. Tanabe, Tetrahedron Lett., 1997, 38, 8727 CrossRef CAS; (c) Y. Yoshida, N. Matsumoto, R. Hamasaki and Y. Tanabe, Tetrahedron Lett., 1999, 40, 4227 CrossRef CAS; (d) R. Hamasaki, S. Funakoshi, T. Misaki and Y. Tanabe, Tetrahedron, 2000, 56, 7423 CrossRef CAS; (e) Y. Tanabe, R. Hamasaki and S. Funakoshi, Chem. Commun., 2001, 1674 RSC; (f) Y. Tanabe, A. Makita, S. Funakoshi, R. Hamasaki and T. Kawakusu, Adv. Synth. Catal., 2002, 344, 507 CrossRef CAS; (g) Y. Tanabe, N. Manta, R. Nagase, T. Misaki, Y. Nishii, M. Sunagawa and A. Sasaki, Adv. Synth. Catal., 2003, 345, 967 CrossRef CAS; (h) A. Iida, S. Nakazawa, T. Okabayashi, A. Horii, T. Misaki and Y. Tanabe, Org. Lett., 2006, 8, 5215 CrossRef CAS.
- (a) Y. Tanabe, N. Matsumoto, S. Funakoshi and N. Manta, Synlett, 2001, 1959 CrossRef CAS; (b) Y. Tanabe, N. Matsumoto, T. Higashi, T. Misaki, T. Itoh, M. Yamamoto, K. Mitarai and Y. Nishii, Tetrahedron, 2002, 58, 8269 CrossRef CAS; (c) Y. Tanabe, K. Mitarai, T. Higashi, T. Misaki and Y. Nishii, Chem. Commun., 2002, 2542 RSC.
- A part of the preliminary result appeared in ref. 6a.
- 1H NMR shifts (ppm) of α-position of two methyl esters and two phenyl esters are as follows. Methyl hexanoate (2.30), phenyl hexanoate (2.55), methyl 2-methyl propanoate (2.56), phenyl 2-methyl propanoate (2.80). This result indicates higher reactivity of phenyl esters.
- T. W. Green and P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 3rd edn, 1999, p. 414 Search PubMed.
- For recent selected works: (a) M. Periasamy, G. Srinivas, G. V. Karunaker and P. Bharathi, Tetrahedron Lett., 1999, 40, 7577 CrossRef CAS; (b) J. C. Adrian, J. L. Barkin, R. J. Fox, J. F. Chick, A. D. Hunter and R. A. Nicklow, J. Org. Chem., 2000, 65, 6264 CrossRef CAS; (c) R. Hayakawa and M. Shimizu, Org. Lett., 2000, 2, 4079 CrossRef CAS; (d) T. Tsuritani, S. Ito, H. Shinokubo and K. Oshima, J. Org. Chem., 2000, 65, 5066 CrossRef CAS; (e) Z. F. Han, H. Yorimitsu, H. Shinokubo and K. Oshima, Tetrahedron Lett., 2000, 41, 4415 CrossRef CAS; (f) A. Kagayama, K. Igarashi, I. Shiina and T. Mukaiyama, Bull. Chem. Soc. Jpn., 2000, 73, 2579 CrossRef CAS.
- The use of other amines, i-Pr2NEt (∼30%), pyridine (trace), PhNMe2 (trace), and Ph2NMe (trace) produced disappointing results.
- (a) Ref. 1b, p. 1230; (b) P. Bako, Á. Szöllõsy, P. Bombicz and L. Tõke, Synlett, 1997, 291 CrossRef CAS.
- (a) N. Kise, K. Tokioka, Y. Aoyama and Y. Matsumura, J. Org. Chem., 1995, 60, 1100 CrossRef CAS; (b) Y. Matsumura, M. Nishimura, H. Hiu, M. Watanabe and N. Kise, J. Org. Chem., 1996, 61, 2809 CrossRef CAS; (c) N. Kise, K. Kumada, Y. Terao and N. Ueda, Tetrahedron, 1998, 54, 2697 CrossRef CAS; (d) V. D. Rao and M. Periasamy, Tetrahedron: Asymmetry, 2000, 11, 1151 CrossRef CAS.
- For example: (a) S. Tamai, H. Ushiroguchi, S. Sano and Y. Nagao, Chem. Lett., 1995, 295 CAS; (b) J. M. Yost, G. Zhou and D. M. Coltart, Org. Lett., 2006, 8, 1503 CrossRef CAS; (c) G. Lalic, A. D. Aloise and M. D. Shair, J. Am. Chem. Soc., 2003, 125, 2852 CrossRef CAS; (d) D. Magdziac, G. Lalic, H. M. Lee, K. C. Fortner, A. D. Aloise and M. D. Shair, J. Am. Chem. Soc., 2005, 127, 7284 CrossRef; (e) The pKa value of α-protons of the thioesters is reported to be approximately 2 units less than that of corresponding esters. F. G. Bordwell and H. E. Fried, J. Org. Chem., 1991, 56, 4218 Search PubMed.
- P. M. Mueller and H. J. Wild, Eur. Pat. Appl., 1992, 479222 Search PubMed; P. M. Mueller and H. J. Wild, Chem. Abstr., 1992, 117, 150865j.
- (a) Y. Yoshida, Y. Sakakura, N. Aso, S. Okada and Y. Tanabe, Tetrahedron, 1999, 55, 2183 CrossRef CAS; (b) J. Morita, H. Nakatsuji, T. Misaki and Y. Tanabe, Green Chem., 2005, 7, 711 RSC and other references cited therein.
- A direct esterification method using p-TsCl–N-methylimidazole also produced successful results (95%). K. Wakasugi, A. Iida, T. Misaki, Y. Nishii and Y. Tanabe, Adv. Synth. Catal., 2003, 345, 1209 Search PubMed.
- J. Canceill and J. Jacques, Bull. Soc. Chim. Fr., 1970, 2180 CAS.
- A. I. Meyers and Y. Yamamoto, J. Am. Chem. Soc., 1981, 103, 4278 CrossRef CAS.
- K. Ishihara, T. Maruyama, M. Mouri, Q. Gao, K. Furuta and H. Yamamoto, Bull. Chem. Soc. Jpn., 1993, 66, 3483 CAS.
- T. Misaki, M. Kurihara and Y. Tanabe, Chem. Commun., 2001, 2478 RSC.
- S. Kiyooka, Y. Kaneko and K. Kume, Tetrahedron Lett., 1992, 33, 4927 CrossRef CAS.
- M. Hirama, D. S. Garvey, L. D.-L. Lu and S. Masamune, Tetrahedron Lett., 1979, 3937 CrossRef.
- R. Annuziata, M. Cinquini, F. Cozzi, P. G. Cozzi and E. Consolandi, Tetrahedron, 1991, 47, 7897 CrossRef.
- L. M. Frost, J. D. Smith and D. J. Berrisford, Tetrahedron Lett., 1999, 40, 2183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/b613544g |
| This journal is © The Royal Society of Chemistry 2007 |