Per(6-guanidino-6-deoxy)cyclodextrins: synthesis, characterisation and binding behaviour toward selected small molecules and DNA†
Nikolaos
Mourtzis
a,
Kyriaki
Eliadou
a,
Chrysie
Aggelidou
a,
Vassiliki
Sophianopoulou
b,
Irene M.
Mavridis
a and
Konstantina
Yannakopoulou
*a
aInstitute of Physical Chemistry, National Center for Scientific Research “Demokritos”, Aghia Paraskevi 15310, Athens, Greece. E-mail: dyanna@chem.demokritos.gr
bInstitute of Biology, National Center for Scientific Research “Demokritos”, Aghia Paraskevi 15310, Athens, Greece
First published on 23rd November 2006
Abstract
Per(6-guanidino-6-deoxy)-cyclodextrins 4a, 4b and 4c are novel derivatives, resulting from homogeneous introduction of the guanidino group at the primary side of α-, β- and γ-cyclodextrins. The products were obtained from the corresponding amino derivatives, as direct guanidinylation of the known bromo-cyclodextrins provided mixtures. The new compounds were fully characterized by NMR spectroscopy and other analytical methods, and their interaction with guest molecules was studied. Strong complexation with 4-nitrophenyl phosphate (NPP) disodium salt was observed (Kbinding ∼5 × 104 M–1), whereas the non-phosphorylated substrate nitrobenzene (NB) formed a very weak complex. 2D ROESY spectra revealed cavity inclusion in both cases, however the orientation of NPP was opposite to that of NB, such that the phosphate group is oriented toward the primary side facing the guanidine groups. The strong affinity of 4 towards the phosphorylated guest suggested that interaction with DNA was possible. The new compounds were found to completely inhibit the migration of ultra pure calf thymus DNA during agarose gel electrophoresis , whereas no effects were observed with guanidine alone or with the plain cyclodextrins . Further, the condensation of DNA into nanoparticles in the presence of 4b was demonstrated by atomic force microscopy, confirming strong electrostatic interaction between the biopolymer and the multicationic products 4. The strong guanidine–phosphate interactions between 4 and DNA were therefore attributed to the clustering of the guanidine groups in the primary area of the cyclodextrin . Cavity effects could not be assessed.
Introduction
Since the publication of the classical paper by Boger, Corcoran and Lehn,1 the chemistry of the cyclic oligosaccharides , cyclodextrins (CDs ), has advanced considerably to provide a variety of well-characterized derivatives.2CDs are cyclic oligosaccharides capable of encapsulating hydrophobic guest molecules in their cavity. Being oligomers of α-D-glucose, CDs are characterised by a set of primary hydroxyl groups directed towards the one side of the macrocyclic structure, and a set of secondary hydroxyl groups directed towards the other (Scheme 1). | ||
| Scheme 1 Representation of the α-, β-, γ-cyclodextrin macrocycles with n = 6, 7, 8, respectively. | ||
There are numerous difficulties associated with the regioselective functionalization of CDs, which involve non-uniform, incomplete or over-substitution, separation of partially substituted products, purification of the products from starting materials and finally comprehensive characterization. Reliable procedures affording cleanly substituted, well-characterized per-6-substituted derivatives continue, therefore, to be in need. Furthermore, the preparation of per-substituted cyclodextrins bearing charged groups has received particular attention3 since the presence of a geometrically defined charged region on the primary or the secondary side may present a significant alternative receptor to the cavity itself. For ionic cyclodextrins purification and identification problems can be considerably larger than outlined above. Several per-(6-anionic-6-deoxy)-derivatives have been reported, which are of interest due to either their unique inclusion properties as drug carriers3c or formation of nanotubes when the secondary side is substituted with complementary cationic groups.3d,e Positively charged derivatives, on the other hand, can act as interesting biomimetic molecules.4 The family of per(6-amino-6-deoxy)cyclodextrins, known for some time,3a,4 constitute a class of compounds that provide both free amine and ammonium groups at neutral pH and behave as biomimetic catalysts.4c Although mono- and di-guanidino βCD derivatives have been prepared5 as probes for interaction with phosphorylated amino acids, the per-guanidino CDs are not known, despite the possibly for more intriguing properties they might present as positively charged, basic oligosaccharides . There is an exceedingly large amount of recent research directed toward the interactions of positively charged macromolecules, mainly polymers,6a–e but also liposomes ,6f and dendrimers6g with DNA, that result in DNA transfection. Some of the most successful cationic polymers6c–e bear cyclodextrin rings linked together by cationic spacer chains. The present work shows that novel per(6-guanidino-6-deoxy)cyclodextrins 4 (Scheme 2) display very strong binding towards phosphorylated and very weak binding towards non-phosphorylated guest molecules. Furthermore, they interact with DNA and actually compact it into nanoparticles, a basic requirement for its entry inside cells.
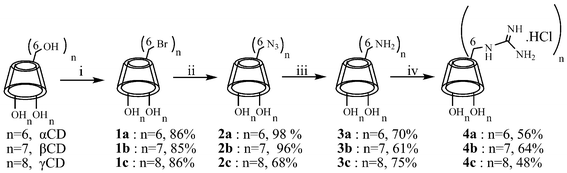 | ||
| Scheme 2 Synthesis of per(6-guanidino-6-deoxy)CDs: (i) DMF, Ph3P, Br2, 60 °C, 45 min, then cyclodextrin addition, 75–80 °C, 18 h, MeONa, MeOH; (ii) DMF, NaN3, NaI, 110 °C, 18 h; (iii) DMF, Ph3P, stir 1.5 h, conc. NH4OH, 18 h; (iv) DMF, 1H-pyrazole-1-carboxamidine·HCl, diisopropylethylamine, 70 °C, 18 h, N2. | ||
Results and discussion
Synthesis and characterization
The synthesis of hexakis-, heptakis-, and octakis(6-guanidino-6-deoxy-)-α-, β- and γ-cyclodextrin, respectively, is shown in Scheme 2. Using literature procedures3a,4a,7 the natural α-, β- and γ-cyclodextrins were first converted to the corresponding per(6-bromo-6-deoxy)-derivatives (1). Direct guanidinylation was considered as an attractive short route to the desired products. Thus, guanidine hydrochloride was converted to free base with either sodium hydroxide or barium hydroxide or sodium methoxide in methanol and allowed to react with compounds 1 in dimethylformamide at either 55–60 °C or 75–80 °C. The results varied, as the substitution was incomplete and several times the major product was the corresponding per(6-chloro-6-deoxy)-derivative,3a along with starting material, whereas the formation of per(3,6-anhydro)-cyclodextrins8a under the basic reaction conditions could not be excluded. The same reaction was attempted starting from guanidine nitrate also without success. Thus, a longer but efficient route was followed (Scheme 2), where compounds 1 were converted to per(6-azido-6-deoxy)-cyclodextrins (2),1,3a,7,8b then to per(6-amino-6-deoxy)-cyclodextrins (3),1,3a,7,8b and finally to per(6-guanidino-6-deoxy)cyclodextrins 4a–c, after treatment of 3 with 1H-pyrazole-1-carboxamidine hydrochloride as the guanylating reagent.5,9 The final products were purified with either chloroform washing (4a) or membrane dialysis (4b, 4c) whereupon small molecules such as reactants and solvents were removed. A very useful indication for the sequential functional group transformations were the chemical shifts of the carbon atoms C6 of the primary side, which changed from ∼62 ppm in the natural CDs to ∼34 ppm in 1a–c, ∼51 ppm in 2a–c, ∼39 ppm in 3a–c and ∼42 ppm in 4a–c.The new products were fully characterised using NMR spectroscopy. The 1H NMR and 13C NMR spectra of 4a, 4b, and 4c in D2O correspond to per-substituted cyclodextrins having C6, C7 and C8 symmetry, respectively (Fig. 1a). The guanidino group protons, absent in D2O, emerged in DMSO-d6 as two broad singlets at 7.8 ppm and 7.2 ppm (the amino group of precursor 3b appeared at 8.24 ppm in DMSO-d6) with a peak area ratio of about 1 : 4, corresponding to (C6-NH) and [-C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)NH2·HCl], respectively. This assignment was based on the 2D 1H–15N correlation spectrum (Fig. 1b), where connectivity of these proton signals with the 15N signals at 78.5 ppm and 72.3 ppm, respectively, was shown. The latter appeared at an area ratio of roughly 1 : 2, therefore correspond to one and two nitrogen atoms, respectively. Moreover, the guanidino protons at 7.8 ppm (C6-NH) J-coupled with the protons H6 and H6′ in COSY spectra in DMSO, verifying substitution on C6 (see also Supporting information† ).
NH)NH2·HCl], respectively. This assignment was based on the 2D 1H–15N correlation spectrum (Fig. 1b), where connectivity of these proton signals with the 15N signals at 78.5 ppm and 72.3 ppm, respectively, was shown. The latter appeared at an area ratio of roughly 1 : 2, therefore correspond to one and two nitrogen atoms, respectively. Moreover, the guanidino protons at 7.8 ppm (C6-NH) J-coupled with the protons H6 and H6′ in COSY spectra in DMSO, verifying substitution on C6 (see also Supporting information† ).
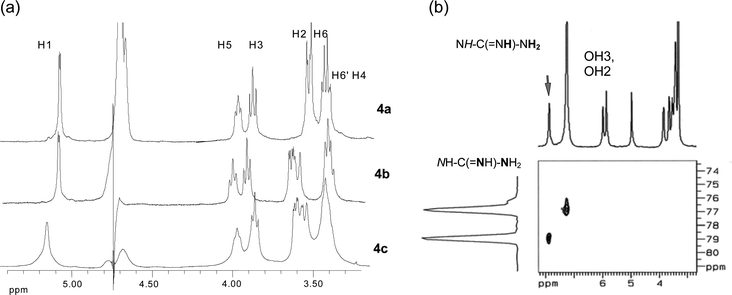 | ||
| Fig. 1 (a) Partial 1H NMR spectra of 4a–c in D2O; (b) 1H–15N correlation spectrum of 4b in DMSO-d6 with the signals of the guanidino group. | ||
Furthermore, electrospray ionization mass spectra revealed the molecular ion in either positive [(MH)+ at m/q 1423] or negative ion mode [(M + Cl)– at m/q 1457], whereas the base peak at m/q 204 was identified as the heptakis protonated molecular ion [(M + 7H+)/7]. Similarly, derivatives, 4a and 4c were characterised in detail. The compounds were isolated as the corresponding hydrochloride salts, as spectrophotometric analyses of chloride ions 10 and elemental analyses showed, holding at least as many Cl– anions as the number of guanidino groups. Treatment of the products with a chloride anion exchanging resin ensured the presence of a constant number of anions in the compounds after each preparation.
Attempts were made to measure the pKa of 4 by monitoring the displacements of the 13C NMR signal of C6 while varying the pH.11 The compounds, however, precipitated at pH 10, therefore the apparent (assuming all ionizations identical) pKa can realistically be placed close to 10, in contrast to the high pKa of guanidine itself (12.5). The products, therefore, are protonated at neutral pH. It has been found that the pKa of the amino cyclodextrins ranges from 7.9 to 8.2,4b,4e much reduced relative to that of the parent amines (e.g.methylamine pKa = 10.67), a feature commonly observed in biological host molecules and in polycationic polymers which catalyse proton transfer reactions. This property is thought to be due to electrostatic through bond effects.12 The pKa values for the guanidino cyclodextrins 4a–c seem to be similarly reduced by at least two units compared to guanidine. Indeed, large pKa shifts are observed for lysine active sites of natural enzymes allowing them to be active at pH 7, where alkylamines, being fully protonated, are unable to act as nucleophiles or general base catalysts.12 The same feature has been demonstrated for amino cyclodextrins , which act as biomimetic catalysts due to cooperativity of cavity binding and the electrostatic effects.4b Interestingly, in the pH range of 6.5–9.5 a complicated 1H NMR spectrum was observed for the amine 3a whereas outside this range the spectrum was a nearly first-order, easily assignable spectrum (Supporting Information† ). The apparent pKa of 3a lies in the pH range where the complex 1H NMR spectra were observed. Apparently, partial protonation of the primary side amino groups resulted in various stable forms of the hexakis(6-amino-6-deoxy)-α-cyclodextrin, separately observable in the NMR spectrum. This NMR behaviour has not been reported previously.1,3a
Binding behaviour of per-guanidino cyclodextrins with nitrobenzene and its 4-phosphate disodium salt substituted analogue
Binding of selected guest molecules into the cavity of the new hosts was examined with 1H NMR spectroscopy. Shielding of the signals of the cavity protons of 4b (Scheme 1), H3 and H5, in the presence of excess nitrobenzene (NB) guest was observed in aqueous solution. Specifically ΔδH3 was 0.081 ppm and ΔδH5 was 0.113 ppm, signifying inclusion. Moreover, strong cross-peaks were observed in the corresponding 2D ROESY spectrum between the protons o- to the nitro group of the guest and H3, H5 and H6 of 4b, and between the m-protons of the guest and H3 of 4b (Fig. 2a), whereas the p-protons did not exhibit any interaction. The above suggest that the guest is enclosed in the cavity and the nitro group is located close to the primary side of 4b. Next, a phosphate substituted nitrobenzene, 4-nitrophenyl phosphate disodium salt hexahydrate (NPP), was examined using 31P NMR in the presence of excess 4b. A large shift in the phosphorous signal was observed (Δδ = 0.185 ppm), compared to that of the guest alone, as well as some peak broadening, that can be ascribed to multiple guanidine–phosphate interactions. Addition of excess guest to 4b, caused its cavity protons to shift appreciably (ΔδH3 = 0.151 ppm, ΔδH5 = 0.364 ppm), showing that the guanidino groups of the host and the phosphate group of the guest interact via cavity insertion of the phenyl part, although external interaction is possible with excess NPP. Indeed, the corresponding 2D ROESY spectrum (Fig. 2b) showed that the protons o- to the nitro group display a stronger cross-peak with the H3 than with H5. In contrast, the protons m- to the nitro group, display a stronger cross-peak with the H5 than with H3. This leads to a binding model where the phosphate group is directed toward the primary side and the nitro group toward the secondary side of 4b Thus inside the cavity of 4b the guests NB and NPP assume opposite orientations.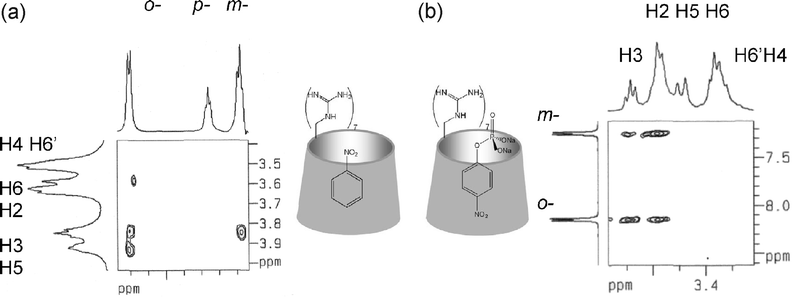 | ||
| Fig. 2 Partial 2D ROESY NMR spectra in D2O of (a) nitrobenzene–4b; (b) 4-nitrophenyl phosphate disodium salt–4b and model representations of the respective inclusion complexes . | ||
Titration of aqueous solutions of 4b with NB and NPP gave a quantitative measure of the strength of binding of the host toward plain or phosphate containing substrates. Thus, the additions of NB into a 1 mM solution of 4b resulted in continuous (therefore fast exchange between formed complex and its free components) shielding of the cavity protons, but the shifts were very small at this concentration, reaching ΔδH3 = 0.011 ppm and ΔδH5 = 0.017 ppm at the 1 : 1 equivalent point. The shielding continued past this point and seemed to curve after 1 : 2 equivalents, indicating guest/host stoichiometry higher than 1. Data fitting using the 1 : 1 binding model13a was unsuccessful, too. It can be concluded, however, based on the very minute shifts, that the binding constant is small, at the limits that can be measured using NMR spectroscopy. In contrast, titration of a 1 mM aqueous solution of 4b with NPP resulted in very large shifts accompanied by signal broadening of the cavity protons, namely ΔδH3 = 0.180 ppm and ΔδH5 = 0.340 ppm at the 1 : 1 equivalent point. Data fitting, attempted despite errors in the shifts due to broad signals, gave an acceptable estimate for the binding constant of 1 : 1 complex, 50460 M–1 ± 9370, with goodness of fit R2 = 0.9368. This value shows very strong binding, a combined effect of cavity inclusion and guanidinium–phosphate interactions. In fact, a binding constant of ∼5 × 104 M–1 corresponds to –ΔG ∼6.4 kcal mol–1, higher than any of the literature reported binding energies of aryl phosphates with A,C or A,D-bis-guanidinium substituted cyclodextrins .4c The presence of seven guanidinium ions in 4b apparently presents a strong advantage toward complexation with the phosphate anion. In conclusion, the strong interactions of the guanidino groups on the primary side of the cyclodextrin with the phosphate group of the guest induce a turnaround in the orientation of the nitro group in NPP, relative to NB, inside the cavity.
The above results show clearly that guanidinylated cyclodextrins bind tightly to phosphorylated substrates. Therefore, investigation of the binding behaviour of 4a–c toward DNA was considered, as a recent report has shown14 that amino cyclodextrins can be used in DNA transfection studies.
Binding behaviour of per-guanidino cyclodextrins with DNA
The influence of compounds 4a–c on double-stranded DNA, was examined using agarose gel electrophoresis and atomic force microscopy (AFM).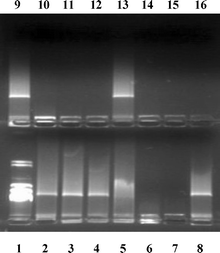 | ||
| Fig. 3 Electrophoresis of calf thymus DNA in a double agarose gel in the presence of 4a–c and control compounds: Lane 1, λHindIII–DNA; Lanes 2 and 9, DNA alone; Lane 3, guanidine hydrochloride; Lane 4, αCD; Lane 8, βCD; Lane 13, γCD; Lanes 5, 6, 7, DNA : 4a mass : charge ratio of 5 : 1, 2 : 1 and 1 : 1, respectively; Lanes 10, 11, 12, DNA : 4b mass : charge ratio of 5 : 1, 2 : 1 and 1 : 1, respectively; Lanes 14, 15, 16, DNA : 4c mass : charge ratio of 5 : 1, 2 : 1 and 1 : 1, respectively. | ||
Interaction with 4a, 4b and 4c is expected to take place through guanidino–phosphate interactions, resulting in modification of the DNA charge and therefore a change in the structure of the macromolecule. This property is augmented as the number of guanidino groups on the CD torus increases from six to eight. Thus, the minimum DNA : compound mass : charge ratio required for the complete inhibition of DNA gel migration is 1 : 1 for 4a, 2 : 1 for 4b and 5 : 1 for 4c. This can be attributed to the fact that a higher local concentration of positively charged guanidino groups results in more efficient charge neutralization of the DNA molecule, resulting in serious restructuring of the macromolecule . This is a clear example of how the clustering of guanidino groups together defines an area for stronger, additive multivalent interactions16 presumably with the phosphate groups of DNA, a feature frequently observed in biological systems. One such system is represented by protamines, small proteins whose role is to pack DNA in sperm heads.17 They are rich (>50%) in arginines, forming sequences of five, six or seven consecutive such residues.18a These proteins, highly hydrophilic and basic, have been found by ESI-MS to bear less charges than anticipated,18b due to electrostatic interactions between successive arginines. Furthermore, protamines interact with the major groove18c,18e through core α-helices,18d,e establishing interactions with the negatively charged phosphate groups. The rigid, guanidino compounds 4a–c bare similarities with the protamine characteristics, since they feature six, seven or eight consecutive guanidino groups clustered in a fixed area of 5–8 Å in diameter (comparable to the diameter of an α helix), therefore it is reasonable to propose similar mode of interaction with DNA.
Finally, comparison of the electrophoretic behaviour of the amino cyclodextrins 3a–c with the guanidines 4a–c showed that even at 1 : 1 DNA : 3b mass : charge ratio complete inhibition of DNA migration was not observed.
 | ||
| Fig. 4 AFM images of plasmid DNA alone (left) and after incubation with 4b (right). | ||
The guanidino cyclodextrins are apparently able to induce condensation of DNA to nanosized particles, a very desirable feature, prerequisite for transporting of DNA through cell membranes. As it has been recently calculated, even a small number of charge neutralizations of DNA, such as that provided by an arginine group, can bend DNA.20 The present work shows that even small, oligocationic molecules, such as 4a–c can be potent in condensing DNA. In a previous report, heptakis(6-amino-6-deoxy)-β-cyclodextrin and several pyridylamino- and imidazolyl-β-cyclodextrins have been shown14 to complex with plasmid DNA, facilitate cell permeation and be effective in DNA transfection requiring, however, large N : P (nitrogen : phosphate) ratios which correspond to cyclodextrin : DNA mass ratios of 100 or higher. For a comparison, we found that a mass ratio of 70 : 1 for heptakis(6-amino-6-deoxy)-β-cyclodextrin : DNA was necessary to achieve mobility inhibition of DNA during electrophoresis . Compounds 4a–c are effective at actual cyclodextrin : DNA mass ratios of 6–1.6, indicating high potency.
The present derivatives are therefore, powerful examples of non-polymeric oligocationic materials that effectively condense DNA, presumably via non-specific guanidine–phosphate interactions. It is suggested that the well defined cyclodextrin shape that enables concentration of many guanidino groups in the same region is responsible for the observed strong effect on DNA. Influence of the cavity, although considered improbable, cannot be assessed. Crystallisation efforts are in progress.
Experimental
Hexakis(6-guanidino-6-deoxy)-α-cyclodextrin hydrochloride (4a)
Hexakis(6-amino-6-deoxy)cyclodextrin (3) as the free base (50 mg, 0.052 mmol), was dispersed in dry dimethylformamide (0.6 ml) and to the mixture 1H-pyrazolecarboxamidine hydrochloride (2.3 eq., 0.72 mmol, 0.11 g) and N,N-diisopropylethylamine (DIPEA) (4.65 eq., 1.45 mmol, 0.20 ml) were added. The whole was stirred at 70 °C for 8 h under a nitrogen atmosphere and then a second addition of the same quantities of 1H-pyrazolecarboxamidine hydrochloride and DIPEA as before, followed. Heating at 70 °C and stirring continued for a further 14 h under nitrogen. Then, diethyl ether (15 ml) was added dropwise and the suspension formed was stirred for 2 h. The solvent was decanted and the collected sticky solid was dissolved in a very small amount of water (0.3 ml). Addition of ethanol resulted in the precipitation of a white substance, which was filtered and dried under vacuum. This precipitate was redissolved in water, the pH was adjusted to 8.5 with sodium hydrogen carbonate and the solution was washed with chloroform (3 × 5 ml), and then treated with Dowex Type I resin (Cl– exchanger) and lyophilized. Yield 56%. 1H NMR (500 MHz, D2O, 298 K): δ 5.04 (d, J = 3.2 Hz, 6H, H1), 3.98 (br m, 6H, H5), 3.92 (t, J = 8.4 Hz, 6H, H3), 3.57 (m, 12H, H6, H2), 3.49 (m, 12H, H4, H6′) ppm; 1H NMR (500 MHz, DMSO, 298 K) δ 7.85 (br s, 6H, C6-NH), 7.18 (br s, 24H, C(NH)-NH2·HCl], 5.66 (d, J = 7.15 Hz, 6H, OH2), 5.51 (br s, 6H, OH3) 4.95 (d, J = 3.1 Hz, 6H, H1), 3.89 (br m, 6H, H5), 3.78 (br t, J = 8.5 Hz, 6H, H3), 3.50–3.40 (m, 12 H, H6, H6′), 3.40–3.30 (br, m, 12H, H2, H4) ppm; 13C NMR (125 MHz, D2O, 298 K) δ 158.22 (-C), 101.76 (C1), 82.76 (C4), 73.10 (C5), 71.76 (C2), 71.42 (C3), 42.75 (C6) ppm; ESI-MS (positive ion mode), calcd: M+ 1219.2. Found: m/q 204.2 ([M + 6H+]/6, 100%) (Found: C, 33.0, H, 6.5, N, 18.2. C42H78O24N18·6HCl·6H2O·C4H6N4 (1H-pyrazolecarboxamidine) requires C, 33.4, H, 6.2, N, 18.6%).
Heptakis(6-guanidino-6-deoxy)-β-cyclodextrin (4b)
The title compound was obtained using heptakis(6-amino-6-deoxy)-β-cyclodextrin (3b) (0.667 g, 0.59 mmol) using the same equivalents of reagents as described above for 4a. A white solid was collected, which was purified in a dialysis membrane (Sigma benzoylated tubing) for 48 h, treated with Dowex Type I resin and lyophilized (64%). 1H NMR (500 MHz, D2O, 300 K) δ 5.20 (d, J = 3.3 Hz, 7H, H1), 4.00 (t, J = 9.7 Hz, 7H, H5), 3.92 (t, J = 9.5 Hz, 7H, H3), 3.65 (dd, J = 3.5 Hz, J = 9.5 Hz, 7H, H2), 3.61 (d, J = 14.9 Hz, 7H, H6), 3.43–3.41 (m, 14H, H4, H6′) ppm; 1H NMR (500 MHz, DMSO-d6, 300 K) δ 7.87 (br, s, 7H, C6-NH), 7.24 [br, s, 21H, C(NH)-NH2], 5.97 (br, s, 7H, OH2), 5.85 (br, s, 7H, OH3) 4.96 (br s, 7H, H1), 3.83 (br, m, 7H, H5), 3.64 (br, m, 7H, H3), 3.53 (m, 7H, H6), 3.44–3.35 (br, m, 21H, H6′, H2, H4) ppm; 13C NMR (125 MHz, D2O, 300 K) δ 158.2 (C), 102.2 (C1), 82.9 (C4), 72.8 (C5), 72.1 (C2), 71.2 (C3), 42.6 (C6) ppm; 15N NMR (50.66 MHz, 1.5 M in D2O, 300 K, D1 = 60 s) δ 78.5 (NH), 72.3 (-NH2) ppm. ESI-MS (positive ion mode), calcd: M+ = 1422.4. Found: m/q 204.3 ([M + 7H+]/7, 100%), 1423 (MH+, 5%). Exact mass (MALDI) for [C49H91O28N21·HCl·Na]+ calcd 1480.6006. Found 1480.7453 (Found: C, 31.0; H, 5.8; N, 15.4. C49H91O28N21·8HCl·9H2O requires C, 31.4; H, 6.3; N, 15.7%).
Octakis(6-guanidino-6-deoxy)-γ-cyclodextrin (4c)
The title compound was obtained using octakis(6-amino-deoxy)-γ-cyclodextrin (3c) (0.561 g, 0.44 mmol) using the same equivalents of reagents as described above for 4a. A white solid was recovered, which was purified in a dialysis membrane (Sigma benzoylated tubing) for 48 h, treated with Dowex Type I resin and either lyophilized or water was evaporated to dryness (48%). 1H NMR (500 MHz, D2O, 300 K) δ 5.14 (br, 8H, H1), 4.05 (m, 8H, H5), 3.90 (t, J = 9.3 Hz, 8H, H3), 3.63 (dd, J = 3.6 Hz, J = 9.8 Hz, 8H, H2), 3.58 (d, J = 14.5 Hz, 8H, H6), 3.54–3.45 (m, 16H, H4, H6′) ppm; 13C NMR (125 MHz, D2O, 300 K), 158.0 (C), 101.0 (C1), 81.0 (C4), 73.0 (C5), 72.9 (C2), 72.8 (C3), 41.0 (C6) ppm; ESI-MS (positive ion mode) calcd: M+ = 1625.6. Found m/q 204.3 ([M + 8H+]/8, 100%), 408 ([M + 4H+]/4, 12%), 543 ([M + 3H+]/3, 17%), 813 ([M + 2H+]/2, 7%); Exact mass (MALDI) calcd for [C56H104O32N24·4HCl·Na]+ = 1791.6213. Found: 1791.6920 (Found: C, 33.9; H, 6.4; N, 16.2. C56H104O32N24·8HCl·5H2O requires C, 33.5; H, 6.1; N, 16.7%).
Agarose gel electrophoresis
Ultrapure calf thymus DNA (∼8.6 MDa, >13 kb, Sigma) (1 µl of a 12.5 µg per 50 µl solution in doubly distilled, autoclaved water) was mixed with increasing volumes (2, 5, 10 µl) of solution (0.1045 mM) of 4a, 4b or 4c in order to achieve a DNA : compound mass : charge ratio of 5 : 1, 2 : 1 and 1 : 1, respectively. Each mixture was diluted with water to a total volume of 20 µl and incubated for approximately 45 min at room temperature. Control samples with guanidine hydrochloride (10 µl of 0.1706 mM solution, DNA : compound mass : charge ratio 1 : 1) and αCD, βCD, γCD (10 µl of 0.1045 mM solutions) were prepared in the same way. After incubation, 2 µl of a dye (0.4% Bromophenol Blue, 67% sucrose) were added to each sample that was then loaded in a 1% agarose gel containing 10 µg ethidium bromide [10 mg ml–1 per 100 ml TAE buffer (40 mM Tris-acetate, 1 mM EDTA)] and electrophoresed, using λHindIII as the molecular weight marker (lane 1). For exact volumes of solutions used, see supporting information.†Atomic force microscopy
Samples were examined using a Digital Instruments Multimide AFM with Nanoscope III controller, operating in Tapping mode. The Si AFM tips, having a typical radius of curvature of 7 nm, and mounted on cantilevers with a resonant frequency of approximately 300 kHz, were supplied by NANOSENSORS. The images had a 512 × 512 pixel resolution with the scan range varying between 1 µm × 1 µm and 6 µm × 6 µm. The tip was operated at a scan rate of 1–3 Hz (lines s–1). AFM images of the plasmid pcDNA3 (5446 bases) alone and in the presence of 4b were collected. 1 µl of pcDNA3 solution (4.4 µg µl–1) was diluted to 100 µl with HEPES–NaOH buffer (pH 7.4) resulting in a 44 ng µl–1 solution. The buffer solution was prepared with ultrapure water (18.2 MΩ) and filtered through a 0.22 µm Millipore filter prior to use. For the DNA condensation studies, 10 µl of pcDNA3 solution were mixed with 10 µl solution (6 × 10–3 M) of 4b and left for incubation for 1 h at room temperature. Then 5 µl of the above mixture were placed on a mica surface and left to dry under a stream of air for 30 min. For the control experiment, 5 µl of pcDNA3 solution were used in the same way.Acknowledgements
The financial support of GSRT programs “Aristeia” (Excellence in the Research Institutes in the frame of articles 4 & 6 of N.2860/00 and EU regulations 1260/99 and 438/01) to K. E., and PRAXE for partial support to N. Mourtzis is appreciated. The NCSR “Demokritos” scholarships to N. M. and C. A. are acknowledged. The authors also thank Ms M. Archimandriti and Dr J. Nolan for help with the AFM measurements, Mr E. Ghika for spectrophotometric chloride determinations, and Dr M. Paravatou and P. Klimentzou for kindly providing the plasmid DNA.References
- R. J. Boger, R. J. Corcoran and J.-M. Lehn, Helv. Chim. Acta, 1978, 61, 2190–2218 CrossRef CAS.
- A. R. Khan, P. Forgo, K. J. Stine and V. T. D'Souza, Chem. Rev., 1998, 98, 1977–1996 CrossRef CAS.
- (a) F. Guillo, B. Hamelin, L. Jullien, J. Canceill, J.-M. Lehn, L. De Robertis and H. Driguez, Bull. Soc. Chim. Fr., 1995, 132, 857–866 CAS; (b) T. Kraus, M. Budesinsky and J. Zavada, Eur. J. Org. Chem., 2000, 3133–3137 CrossRef CAS; (c) A. Bom, M. Bradley, K. Cameron, J. K. Clark, J. van Egmond, H. Feilden, E. J. MacLean, A. W. Muir, R. Palin, D. C. Rees and M.-Q. Zhang, Angew. Chem., Int. Ed., 2002, 41, 265–270 CrossRef CAS; (d) T. Kraus, M. Budesinsky, I. Cisarova and J. Zavada, Angew. Chem., Int. Ed., 2002, 41, 1715–1717 CrossRef CAS; (e) T. Kraus, M. Budesinsky and J. Zavada, J. Org. Chem., 2001, 66, 4595–4600 CrossRef CAS.
- (a) D. Vizitiu, C. S. Walkinshaw, B. I. Gorin and G. R. J. Thatcher, J. Org. Chem., 1997, 62, 8760–8766 CrossRef; (b) C. G. Ferguson and G. R. J. Thatcher, Org. Lett., 1999, 1, 829–832 CrossRef CAS; (c) S. L. Hauser, E. W. Johanson, H. P. Green and P. J. Smith, Org. Lett., 2000, 2, 3575–3578 CrossRef CAS; (d) S. L. Hauser, E. S. Cotner and P. J. Smith, Tetrahedron Lett., 1999, 40, 2865–2866 CrossRef CAS; (e) W. Tagaki, K. Yano, K. Yamanaka, H. Yamamoto and T. Miyasaka, Tetrahedron Lett., 1990, 31, 3897–3900 CrossRef CAS.
- E. S. Cotner and P. J. Smith, J. Org. Chem., 1998, 63, 1737–1739 CrossRef CAS.
- (a) W. T. Godbay, K. K. Wu and A. G. Mikos, Biomaterials, 2001, 22, 471–480 CrossRef; (b) P. Dubruel, B. Christiaens, B. Vanloo, K. Bracke, M. Rosseneu, J. Vandekerchove and E. Schaht, Eur. J. Pharm. Sci., 2003, 18, 211–220 CrossRef CAS and references therein; (c) H. Gonzalez, S. J. Hwang and M. E. Davis, Bioconjugate Chem., 1999, 10, 1068–1074 CrossRef CAS; (d) S. J. Hwang, N. C. Bellocq and M. E. Davis, Bioconjugate Chem., 2001, 12, 280–290 CrossRef CAS; (e) S. R. Popielarski, S. Mishra and M. E. Davis, Bioconjugate Chem., 2003, 14, 672–678 CrossRef CAS; (f) R. Banerjee, V. Y. Mahidhar and A. Chaudhuri, J. Med. Chem., 2001, 44, 4176–4185 CrossRef CAS and references cited; (g) F. Kihara, H. Arima, T. Tsutsumi, F. Hirayama and K. Uekama, Bioconjugate Chem., 2003, 14, 342–350 CrossRef CAS.
- (a) H. H. Baer, A. V. Berenguel, Y. Y. Shu, J. Defaye, A. Gadelle and F. S. Gonzalez, Carbohydr. Res., 1992, 228, 307–314 CrossRef CAS; (b) A. Gadelle and J. Defaye, Angew. Chem., Int. Ed. Engl., 1991, 30, 78–80 CrossRef; (c) K. Takeo, T. Sumimoto and T. Kuge, Staerke, 1974, 26(4), 111–115 Search PubMed; (d) M. N. Berberan-Santos, J. Canceill, J.-C. Brochon, L. Jullien, J.-M. Lehn, J. Pouget, P. Tauc and B. Valeur, J. Am. Chem. Soc., 1992, 114, 6427–6436 CrossRef CAS; (e) B. A. Gorin, R. J. Riopelle and G. R. J. Thatcher, Tetrahedron Lett., 1996, 37(27), 4647–4650 CrossRef CAS.
- (a) P. R. Ashton, R. Ellwood, I. Staton and J. F. Stoddart, J. Org. Chem., 1991, 56, 7274–7280 CrossRef CAS; (b) P. R. Ashton, R. Koeniger, J. F. Stoddart, D. Alker and V. D. Harding, J. Org. Chem., 1996, 61, 903–908 CrossRef CAS.
- D. A. Powell, P. D. Ramsden and R. A. Batey, J. Org. Chem., 2003, 68, 2300–2309 CrossRef CAS.
- T. M. Florence and Y. J. Farrar, Anal. Chim. Acta, 1971, 54, 373–377 CrossRef CAS.
- (a) C. S. Handloser, M. R. Chakrabarty and M. W. Mosher, J. Chem. Educ., 1973, 50, 510–511 CrossRef CAS; (b) H. Astroem, E. Limen and R. Stroemberg, J. Am. Chem. Soc., 2004, 126, 14710–14714 CrossRef.
- F. Hollfelder, A. J. Kirby and D. S. Tawfik, J. Am. Chem. Soc., 1997, 119, 9578–9579 CrossRef CAS.
- (a) K. Eliadou, K. Yannakopoulou, A. Rontoyianni and I. M. Mavridis, J. Org. Chem., 1999, 64, 6217–6226 CrossRef CAS.
- S.-A. Cryan, A. Holohan, R. Donohue, R. Darcy and C. M. O'Driscoll, Eur. J. Pharm. Sci., 2004, 21, 625–633 CrossRef CAS.
- Since DNA and the prepared cyclodextrin derivatives are both charged, for each molecule the mass : charge ratio was calculated. For the compounds 4a–4c the calculation of charges was based on the spectrophotometric analysis of chloride ions and the elemental analysis results, and therefore the molecular weight was accordingly corrected by adding the appropriate number of HCl molecules to the cyclodextrin formula in each case. For DNA the average molecular weight was taken as 330 per base.
- (a) D. A. Fulton, S. J. Cantrill and J. F. Stoddart, J. Org. Chem., 2002, 67, 7968–7981 CrossRef CAS; (b) M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- L. Stryer, Biochemistry, W. H. Freeman & Co, New York, 3rd edn, 1988, p. 830 Search PubMed.
- (a) P. Sautiere, D. Belaiche, A. Martinage and M. Loir, Eur. J. Biochem., 1984, 144, 121–125 CrossRef CAS; (b) P. Schindler, F. Bitsch, K. Klarskov, P. Roepstorff, G. Briand, D. Wouters-Tyrou and A. Van Dorsselaer, Eur. J. Biochem., 1991, 195, 621–629 CrossRef CAS; (c) I. Fita, J. L. Campos, L. C. Puigjaner and J. A. Subirana, J. Mol. Biol., 1983, 167(1), 157–177 CAS; (d) R. W. Warrant and S. H. Kim, Nature, 1978, 271(5641), 130–135 CAS; (e) A. Kagemoto, N. Fujita, K. Ueno and Y. Baba, J. Phys. Chem., 1995, 99, 424–430 CrossRef CAS.
- (a) J. A. Bordelon, K. J. Feierabend, S. A. Siddiqui, L. L. Wright and J. T. Petty, J. Phys. Chem. B, 2002, 106, 4838–4838 CrossRef CAS; (b) C. Rivetti, M. Guthold and C. Bustamante, J. Mol. Biol., 1996, 264, 919–932 CrossRef CAS.
- G. S. Manning, J. Am. Chem. Soc., 2003, 125, 15087 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental data, melting points, spectroscopic characterisation (NMR, IR) of all known compounds 1–3, 1H NMR spectra of 3a–c, 2D NMR spectra of 4a–c, mass spectra and volumes of solutions used in electrophoresis . See DOI: 10.1039/b614899a |
| This journal is © The Royal Society of Chemistry 2007 |