A new strategy for the synthesis of taurine derivatives using the ‘safety-catch’ principle for the protection of sulfonic acids
Received 2nd October 2006, Accepted 8th November 2006
First published on 24th November 2006
Abstract
The safety-catch principle has been applied for the development of a new method for protecting sulfonic acids. 2,2-Dimethylsuccinic acid was reduced to 2,2-dimethylbutane-1,4-diol, which was selectively silylated to give 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutan-1-ol. Reaction of the latter compound with 2-chloroethanesulfonyl chloride in the presence of triethylamine afforded 4-(tert-butyldiphenylsilyloxy)-2,2-dimethylbutyl ethenesulfonate directly. The ethenesulfonate underwent Michael-type addition with secondary amines to give protected derivatives of taurine (2-aminoethanesulfonic acid). Deprotection was achieved on treatment with tetrabutylammonium fluoride, whereby cleavage of the silicon–oxygen bond led to an intermediate alkoxide that immediately cyclised to 2,2-dimethyltetrahydrofuran with liberation of a sulfonate. Pure sulfonic acids were obtained from the crude product by ion exchange chromatography on a strongly basic resin, which was eluted with aqueous acetic acid. The method developed should be generally applicable to the protection of sulfonic acids and is amenable to a multiparallel format.
Introduction
In a research programme aimed at synthesising potential inhibitors of the histone acetyltransferase enzyme called Tip60 (Tat interacting protein),1 we required a small molecule library in which all of the members terminated with an N,N-disubstituted 2-aminoethylsulfonate, i.e. derivatives of taurine (2-aminoethanesulfonic acid). There is substantial interest in taurine and its derivatives because of the uncertain, but probable important role of this abundant amino acid in human and animal cells.2 The sulfonate moiety can also be used as a surrogate for carboxylate and phosphate in drug development.3 Sulfonate groups are key components of antagonists for P2 purinergic receptors4 and follicle stimulating hormone.5 The wider application of sulfonates in medicinal chemistry may have been frustrated by a lack of satisfactory methods for the masking of this polar, strongly acidic group. Sulfonic acids have been protected as sulfonate esters [isopropyl,5,6 isobutyl,7 neopentyl8m-nitrophenoxy,4 pentafluorophenyl9 or solid supported benzyl (e.g. Wang resin10)], as sulfonamides with secondary amines11 and by ‘non-covalent’ masking with a hydrophobic ammonium species.12 The photolabile 2,5-dimethoxyphenacyl group has also been employed for the protection of sulfonic acids.13 Many of these methods employed acidic (e.g. trifluoroacetic acid)8a or basic conditions (e.g. 2 M NaOH at 70 °C)4 in the cleavage step, which may be unsuitable for some target molecules. We describe a protecting strategy for sulfonic acids in which the versatile, reactive intermediate 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutyl ethenesulfonate 1 reacts in high yields with different secondary amines (e.g. morpholine 2a) to give an adduct (e.g. 2-morpholin-4-yl-ethanesulfonic acid 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethyl-butyl ester 3a), which can be cleaved to the corresponding sulfonic acid (e.g. 2-morpholin-4-yl-ethanesulfonic acid 4a) rapidly and in high yield at room temperature using tetrabutylammonium fluoride (TBAF) in THF. The resulting sulfonic acids are easily purified by ion exchange chromatography. This strategy is an application of Kenner's ‘safety-catch’ principle,14,15 whereby a labile intermediate is released in situ by removal of a relatively robust protecting group.Results and discussion
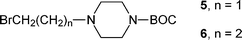
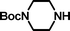
Initially, we planned to introduce the sulfonic acid group in the last step of a synthetic sequence using sulfite (Na2SO3) to displace a bromo group. To this end, N-tert-butyloxycarbonylpiperazine was alkylated with 1,2-dibromoethane and 1,3-dibromopropane giving 5 and 6, respectively. However, the yields of these molecules, which were required for further chain extension, were only 5% (5) and 41% (6), respectively.
We then conceived an alternative strategy based on a protected common intermediate ethenesulfonate, 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutyl ethenesulfonate 1, which could be reacted with a variety of amines prior to deprotection. Thus, reaction of the mono-silyl protected alcohol 7 with commercially available 2-chloroethanesulfonyl chloride gave ethenesulfonate 116 in good yield (Scheme 1). The alcohol 7 was readily prepared by reduction of 2,2-dimethylsuccinic acid 8 with lithium aluminium hydride to afford 2,2-dimethylbutane-1,4-diol 9, which was selectively silylated17 with tert-butyldiphenylsilyl chloride (TBDPS-Cl) at its less hindered hydroxyl group.
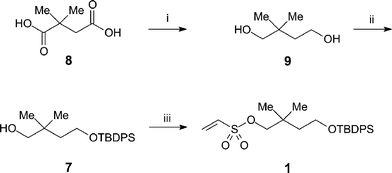 |
| | Scheme 1 Synthesis of 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutyl ethenesulfonate 1. Reagents and conditions: (i) LiAlH4, ether, reflux, 85%; (ii) TBDPS-Cl, imidazole, DMF, rt, 79%; (iii) 2-chloroethanesulfonyl chloride, Et3N, DCM, rt, 93%. | |
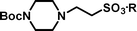
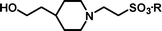
Michael-type coupling of ethenesulfonate 1 with secondary amines R1R2NH 2a–2h occurred smoothly to give adducts 3a–3h, essentially as previously described.18 Deprotection of the adducts with tetrabutylammonium fluoride (TBAF) liberated the desired sulfonic acids (4a–4h). Cleavage of the Si–O bond releases an alkoxide that immediately effects an intramolecular displacement of the sulfonate moiety. Besides providing a neopentyl-like group already known to be advantageous for protection of sulfonic acids, the gem-dimethyl group offers two advantages in this strategy. It ensures regioselective silylation of the intermediate diol 9 and enhances the rate of cyclisation of the desilylated intermediate by a ‘Thorpe–Ingold effect’.19 The deprotection reactions were performed with a small excess (0.1–0.2 equiv.) of TBAF in THF to afford a mixture containing tetrabutylammonium sulfonates, TBDPS-F and volatile 2,2-dimethyltetrahydrofuran. Evaporation removed the dimethyltetrahydrofuran (bp 90 °C at 740 mmHg20), whilst extraction with ethyl acetate removed TBDPS-F. Finally, purification of the tetrabutylammonium sulfonates by anion exchange chromatography and elution with aqueous acetic acid as eluent, afforded highly pure sulfonic acids (Scheme 2). The data in Table 1 demonstrates the efficiency of the coupling reactions of 1 with secondary amines 2a–2h, as well as the deprotection and the purification of the sulfonic acids, which were all obtained in good purity, as determined by 1H NMR and combustion analyses.
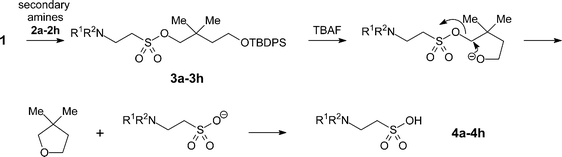 |
| | Scheme 2 Coupling of secondary amines 2a–2h to ethenesulfonate 1 and deprotection of adducts 3a–3h to sulfonic acids 4a–4h. | |
Table 1
Previously, sulfonic acids have been protected with the ‘NeoN-B’ group 108b that was removed by treatment with trifluoroacetic acid, followed by neutralisation. The liberated amino group effected an intramolecular cyclisation releasing the sulfonate in an analogous manner to that described here. However, the synthesis of the alcohol precursor of NeoN-B is lengthy and only one example of its use was described without full experimental details. Another example of this theme is the protection of phenols as carbamates that are cleaved by intramolecular attack by a hydroxyl function that is released from a tri-isopropylsilyl ether by TBAF attack.21
The methods described in this paper should be applicable to the synthesis of wide range of sulfonic acids. The chemistry is amenable to scale-up and can be performed in a multiparallel synthesis format. The methodology described could also be extended, in principle, to the protection of sulfate mono-esters.22
Experimental
General procedures
Chemicals were purchased from the Aldrich Chemical Company. THF was freshly distilled from sodium–benzophenone. Other anhydrous solvents were obtained from Aldrich in SureSeal™ bottles. Triethylamine was distilled from calcium hydride.Thin layer chromatography (TLC) was performed using Merck silica gel 60F254 pre-coated on aluminium sheets. Medium pressure (‘medium pressure’) chromatography was carried out using Davisil silica gel (40–63 µm). For the ion exchange chromatography the strongly basic anion exchange resin Dowex Monosphere 550 A (OH) (capacity 1.2 meq cm−3) from Sigma-Aldrich was used.
1H and 13C nuclear magnetic resonance (NMR) spectra were obtained using a Bruker Spectrospin AC 300 E (1H at 300 MHz or 13C at 75 MHz) or a JEOL JNM-LA500 spectrometer (1H at 500 MHz or 13C at 125 MHz). Chemical shifts are reported in parts per million using residual solvent peaks as internal standards. Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br s (broad signal) or combinations thereof. LC-MS spectra were obtained using a Micromass Platform instrument running in positive or negative ion electrospray mode. IR spectra were recorded on a Bio-Rad FTS 3000MX diamond ATR as a neat sample.
Melting points were determined using a Stuart Scientific SMP3 apparatus and are uncorrected.
General procedure A. For the synthesis of the piperazine-derivatives 5 and 6, tert-butyl piperazine-1-carboxylate, di-isopropylethylamine and the appropriate dibromoalkane were heated at reflux with stirring in anhydrous dichloromethane for at least 2–4 d. After removal of the solvent, the white solid was extracted with ethyl acetate. Unreacted tert-butyl piperazine-1-carboxylate was recovered by filtration and the filtrate was concentrated to give a residue that was purified by medium pressure chromatography.
General procedure B. The ethenesulfonate ester 1 was taken up in dichloromethane and the secondary amine (2–4 equiv., except morpholine) was added to the mixture. After stirring overnight at room temperature, the solvent was removed and the residue was purified by medium pressure chromatography using acetone–hexane as eluent.
General procedure C for the cleavage of the sulfonate protecting group. The sulfonate ester (one of 3a–3h) in THF was added to TBAF in THF and the mixture was stirred at room temperature for 2 d. After extraction with ethyl acetate, the organic phase was washed with water (3×) and the combined aqueous extracts were concentrated. The residual sulfonic acid (one of 4a–4h) containing tetrabutylammonium species was purified by ion exchange chromatography. Dowex Monosphere 550 A (OH) resin (∼5–7 cm3 wet) was flushed with deionised water. A 12.5% (w/v) solution of ammonia in deionised water was passed through the column until the eluent was basic. The resin was rinsed with deionised water until the eluent was neutral and the sulfonic acid 4a–4h in deionised water was delivered to the column. After elution with water to remove Bu4NOH (basic pH), the resin was eluted with 20% (w/v) aqueous acetic acid (400 cm3; there was eventually a distinct lightening in colour of the resin). The solvent was removed and the sulfonic acid 4a–4h was dried at 40–50 °C in vacuo.
tert-Butyl 4-(2-bromoethyl)-piperazine-1-carboxylate (5). Following the general procedure A, the reaction of tert-butyl piperazine-1-carboxylate (505 mg, 2.71 mmol), 1,2-dibromoethane (1.0 cm3, 11.6 mmol) and di-isopropylethylamine (0.50 cm3, 2.88 mmol) in dichloromethane (25 cm3) afforded, after purification by medium pressure chromatography (elution with 15% petrol–ethyl acetate) compound 5 (42 mg, 5%) as a light yellow solid. δH (300 MHz, CDCl3) 1.41 (9 H, s, C(CH3)3), 2.41 (4 H, t, J 4.9, Pip3,5), 2.68 (2 H, t, J 6.9, BrCH2CH2N), 3.39 (4 H, t, J 5.1, Pip2,6), 3.55 (2 H, t, J 6.9, BrCH2CH2N) [not further characterised].
tert-Butyl 4-(3-bromopropyl)-piperazine-1-carboxylate (6). Following the general procedure A, the reaction of tert-butyl piperazine-1-carboxylate (519 mg, 2.79 mmol), 1,3-dibromopropane (1 cm3, 9.85 mmol) and di-isopropylethylamine (0.50 cm3, 2.88 mmol) in dichloromethane (25 cm3) afforded, after purification by medium pressure chromatography (elution with 15% petrol–ethyl acetate) compound 6 (348 mg, 41%) as a light yellow solid, mp 118–121 °C. δH (300 MHz, CDCl3) 1.41 (9 H, s), 1.99 (2 H, t, J 6.8), 2.30–2.40 (4 H, br s), 2.45 (2 H, t, J 6.9), 3.33–3.45 (6 H, m); δC (125.7 MHz, CDCl3) 28.8, 30.0, 32.0, 43.4, 53.4, 56.8, 80.2, 155.1; HRMS (EI+) C12H23BrN2O2 requires 306.0943; found 306.0944.
2,2-Dimethylbutane-1,4-diol (9). A solution of 2,2-dimethylsuccinic acid 8 (4.77 g, 32.6 mmol) in diethyl ether (75 cm3) was added dropwise to a 1 M solution of LiAlH4 in diethyl ether (108 cm3, 0.108 mol). The mixture was heated at reflux for 17 h and quenched carefully with 4 cm3 water, 4 cm3 15% NaOH and 12 cm3 water. After stirring for 30 min, the slurry was filtered and the inorganic salts were extracted three times with boiling diethyl ether. The combined ethereal extracts were dried first with MgSO4 and twice with Na2SO4 and concentrated in vacuo. The resulting colourless viscous oil 9 (3.27 g, 85%) was used without further purification. δH (300 MHz, CDCl3) 0.84 (6 H, s, (CH3)2C), 1.5 (2 H, t, J 5.6, 3-H), 3.27 (2 H, s, 1-H), 3.43 (2 H, s, 2 × OH), 3.64 (2 H, t, J 5.6, 4-H); δC (75.5 MHz, CDCl3) 25.4 (C(CH3)2), 35.3 (C(CH3)2), 43.2 (CH2CH2OH), 59.4 (CH2OH), 71.9 (C(CH3)3CH2OH).
4-(tert-Butyldiphenylsilanyloxy)-2,2-dimethylbutan-1-ol (7). To a solution of the diol 9 (7.39 g, 62.5 mmol) in DMF (100 cm3) was added imidazole (8.05 g, 118.2 mmol, 1.9 equiv.) and tert-butylchlorodiphenylsilane (16.09 g, 58.5 mmol, 0.94 equiv.). After stirring at room temperature for 60 h, the reaction was quenched with saturated aqueous NaHCO3 (50 cm3). The DMF was removed and the residue was extracted with diethyl ether (5×) and ethyl acetate (1×). The combined organic extracts were dried over Na2SO4, filtered and the solvent was removed. The crude product was purified by medium pressure chromatography (elution with 15% acetone–hexane) to give the title compound (16.4 g, 79%) as a colourless oil (Found: C, 73.9; H, 9.1. C22H32O2Si requires C, 74.1; H 9.1%); δH (300 MHz, CDCl3) 0.80 (6 H, s, (CH3)2C), 0.98 (9 H, s, (CH3)3C), 1.47 (2 H, t, J 5.5, 3-H), 3.3 (2 H, s, 1-H), 3.64 (2 H, t, J 5.5, 4-H), 7.3–7.4 (6 H, m, Ph), 7.59–7.63 (4 H, m, Ph); δC (75.5 MHz, CDCl3) 19.4 (C(CH3)3), 25.1 (C(CH3)3), 27.3 (C(CH3)2), 35.3 (2-C), 42.3 (3-C), 61.5 (4-C), 72.2 (1-C), 128.1, 130.0, 134.0, 136.0.1,4-Di-(tert-butyldiphenylsilanyloxy)-2,2-dimethyl-butane (2.76 g, 8%) was also obtained. δH (300 MHz, CDCl3) 0.77 (6 H, s, (CH3)2C), 0.95 (9 H, s, (CH3)3), 0.96 (9 H, s, (CH3)3), 1.56 (2 H, t, J 7.3, 3-H), 3.2 (2 H, s, 1-H), 3.64 (2 H, t, J 7.3, 4-H), 7.3–7.4 (12 H, m, Ph), 7.5–7.6 (8 H, m, Ph); δC (75.5 MHz, CDCl3) 19.5 (C(CH3)3), 19.8 (C(CH3)3), 24.8, 27.2, 27.3, 35.4, 41.8 (C(CH3)3CH2), 61.4 (C(CH3)3CH2CH2), 73.1 (CH2O), 127.9, 128.0, 129.9, 134.2, 134.5, 136.0, 136.1.
Ethenesulfonic acid 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutyl ester (1). To a stirred solution of 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutan-1-ol 7 (6.55 g, 18.37 mmol) in dichloromethane (250 cm3) cooled in an ice-bath were added 2-chloroethanesulfonyl chloride (6.8 g, 41.8 mmol) followed by anhydrous triethylamine (14 cm3, ∼5.2 equiv.). After stirring overnight at room temperature the brown solution was washed with 10% (w/v) aqueous Na2CO3 (1×) and water (2×). The organic layer was dried and the solvent was removed. The residue was purified by medium pressure chromatography on silica using 10% acetone–hexane as eluent, to give the title compound (7.59 g, 93%) as a viscous, yellow oil (Found: C, 64.4; H, 7.6. C24H34O4SSi requires C, 64.5; H, 7.67%); δH (300 MHz, CDCl3) 0.86 (6 H, s, (CH3)2C), 0.99 (9 H, s, (CH3)3C), 1.5 (2 H, t, J 6.6), 3.63 (2 H, t, J 6.6), 3.78 (2 H, s, OCH2), 5.97 (1 H, dd, J 0.8, 8.8, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 6.24–6.40 (2 H, m, CHCH), 7.29–7.40 (m, 6 H, Ph), 7.57–7.60 (m, 4 H, Ph); δC (75.5 MHz, CDCl3) 19.5, 24.7, 27.4, 34.2, 41.7, 60.9, 79.0, 128.1, 129.5, 130.0, 133.4, 134.3, 136.0.
CH), 6.24–6.40 (2 H, m, CHCH), 7.29–7.40 (m, 6 H, Ph), 7.57–7.60 (m, 4 H, Ph); δC (75.5 MHz, CDCl3) 19.5, 24.7, 27.4, 34.2, 41.7, 60.9, 79.0, 128.1, 129.5, 130.0, 133.4, 134.3, 136.0. 2-Morpholin-4-yl-ethanesulfonic acid 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutyl ester (3a). Following the general procedure B, reaction of ethenesulfonate ester 1 (107 mg, 0.24 mmol) with morpholine (388 mg, 4.4 mmol) in dichloromethane (50 cm3) afforded, after purification by medium pressure chromatography (30% acetone–hexane) the title compound (104 mg, 81%) as a colourless oil (Found: C, 62.9; H, 8.2; N 2.62. C28H43NO5SSi requires C, 63.0; H 8.1; N 2.6%); νmax (film)/cm−1 1354, 1167, 1109, 956, 820, 738 and 702; δH (300 MHz, CDCl3) 0.90 (6 H, s, (CH3)2C), 0.97 (9 H, s, (CH3)3C), 1.55 (2 H, t, J 6.7), 2.37–2.41 (4 H, m), 2.79 (2 H, dd, J 6.4, 8.7), 3.20 (2 H, dd, J 6.4, 8.7), 3.60–3.63 (4 H, m), 3.68 (2 H, t, J 6.7), 3.92 (2 H, s), 7.3–7.39 (6 H, m, Ph), 7.57–7.61 (4 H, m, Ph); δC (75.5 MHz, CDCl3) 19.5 (C(CH3)3), 24.7 (C(CH3)3), 27.3 (C(CH3)2), 34.2, 41.7 (C(CH3)3CH2), 48.4 (NCH2CH2SO2), 52.7 (NCH2CH2SO2), 53.7 (2 × morpholine-C), 60.9 (C(CH3)3CH2CH2), 67.1, 78.0 (CH2O), 128.0, 130.0, 134.3, 135.9.
tert-Butyl 4-{2-[4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutoxysulfonyl]-ethyl}-piperazine-1-carboxylate (3b). Following the general procedure B, reaction of ethenesulfonate ester 1 (733 mg, 1.64 mmol) with tert-butyl piperazine-1-carboxylate (600 mg, 3.22 mmol, in 8 cm3 dichloromethane) in dichloromethane (50 cm3) afforded, after purification by medium pressure chromatography (15% acetone–hexane), the title compound (985 mg, 95%) as a light yellow oil (Found: C, 62.8; H, 8.4; N, 4.4. C33H52N2O6SSi requires C, 62.6; H, 8.3; N, 4.4%); νmax (film)/cm−1 1691, 1358, 1247, 1164, 1107, 1001, 955, 823, 738 and 702; δH (300 MHz, CDCl3) 0.89 (6 H, s, (CH3)2C), 0.97 (9 H, s, (CH3)3C), 1.39 (9 H, s, (CH3)3C), 1.54 (2 H, t, J 6.7), 2.33 (4 H, br s), 2.76–2.81 (2 H, m), 3.15–3.20 (2 H, m), 3.35 (4 H, br s), 3.67 (2 H, t, J 6.7), 3.90 (2 H, s), 7.29–7.39 (6 H, m, Ph), 7.57–7.60 (4 H, m, Ph); δC (75.46 MHz, CDCl3) 19.5, 24.6, 27.3, 28.8, 34.2, 41.7, 44.0, 48.5, 52.3, 53.1, 60.8, 78.0, 80.1 128.0, 130.0, 134.2, 135.9, 155.0.
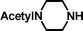
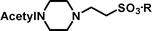
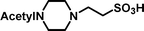
2-(4-Acetylpiperazin-1-yl)-ethanesulfonic acid 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutyl ester (3c). Following the general procedure B, reaction of ethenesulfonate ester 1 (738 mg, 1.65 mmol) with N-acetylpiperazine (716 mg, 5.6 mmol) in dichloromethane (50 cm3) afforded, after purification by medium pressure chromatography (40% acetone–hexane), the title compound (806 mg, 85%) as a light yellow oil (Found: C, 62.5; H, 8.2; N 4.4. C30H46N2O5SSi requires C, 62.7; H, 8.1; N, 4.9%); νmax (film)/cm−1 1636, 1427, 1355, 1165, 1105, 996, 953, 822, 739 and 702; δH (300 MHz, CDCl3) 0.90 (6 H, s, (CH3)2C), 0.97 (9 H, s, (CH3)3C), 1.53 (2 H, t, J 6.7), 2.0 (3 H, s, CH3), 2.35–2.40 (4 H, m), 2.80 (2 H, dd, J 6.4, 8.5), 3.18 (2 H, m), 3.37 (2 H, dd, J 6.6, 11.7), 3.52–3.55 (2 H, m), 3.66 (2 H, t, J 6.6), 3.90 (2 H, s), 7.29–7.39 (6 H, m, Ph), 7.57–7.61 (4 H, m, Ph); δC (75.46 MHz, CDCl3) 19.5, 21.3, 24.6, 27.3, 29.7, 34.2, 41.7, 48.5, 52.1, 60.8, 78.0, 128.0, 130.0, 134.3, 135.9, 169.1.
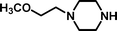
2-[4-(2-Methoxyethyl)-piperazin-1-yl]-ethanesulfonic acid 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutyl ester (3d). Following the general procedure B, reaction of ethenesulfonate ester 1 (720 mg, 1.61 mmol) with 1-(2-methoxyethyl)-piperazine (611 mg, 4.2 mmol) in dichloromethane (50 cm3) afforded, after purification by medium pressure chromatography (30% acetone–hexane), the title compound (889 mg, 93%) as an oil (Found: C, 62.6.; H, 8.6; N, 4.6. C31H50N2O5SSi requires C, 63.0; H, 8.5; N, 4.7%); νmax (film)/cm−1 1354, 1165, 1105, 956, 8.22, 738 and 702; δH (300 MHz, CDCl3) 0.89 (6 H, s, (CH3)2C), 0.97 (9 H, s, (CH3)3C), 1.52 (2 H, t, J 6.7), 2.47–2.82 (9 H, m), 2.79 (2 H, dd, J 6.4, 8.9), 3.17 (2 H, dd, J 6.4, 8.9), 3.28 (3 H, s), 3.44 (2 H, m), 3.65 (2 H, t, J 6.7), 3.88 (2 H, s), 7.29–7.39 (6 H, m, Ph), 7.57–7.60 (4 H, m, Ph); δC (75.46 MHz, CDCl3) 19.5, 24.6, 27.3, 34.2, 41.7, 48.5, 52.2, 53.2, 53.8, 58.1, 59.0, 60.8, 70.9, 78.0, 128.0, 130.0, 134.3, 135.9.
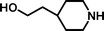
2-[4-(2-Hydroxyethyl)-piperidin-1-yl]-ethanesulfonic acid 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutyl ester (3e). Following the general procedure B, reaction of ethenesulfonate ester 1 (767 mg, 1.72 mmol) with 4-(2-hydroxyethyl)-piperidine (579 mg, 4.5 mmol) in dichloromethane (50 cm3) afforded, after purification by medium pressure chromatography (30% acetone–hexane), the title compound (836 mg, 84%) as a light yellow oil (Found: C, 64.0.; H, 8.6; N, 2.3. C31H49NO5SSi requires C, 64.7; H, 8.6; N, 2.4%); νmax (film)/cm−1 1352, 1165, 1103, 955, 737 and 702; δH (300 MHz, CDCl3) 0.89 (6 H, s, (CH3)2C), 0.97 (9 H, s, (CH3)3C), 1.17–1.65 (9 H, m), 1.98 (2 H, br t), 2.77 (4 H, m), 3.20 (2 H, m), 3.59–3.68 (4 H, m), 3.89 (2 H, s), 7.32–7.39 (6 H, m, Ph), 7.58–7.60 (4 H, m, Ph); δC (75.46 MHz, CDCl3) 19.5, 24.6, 27.3, 32.7, 34.2, 39.7, 41.7, 48.7, 52.5, 54.0, 60.8, 78.0, 128.0, 130.0, 134.3, 135.9; HRMS (EI+) C31H49NO5SSi requires 575.3101, found 575.3095.
2-Pyrrolidin-1-yl-ethanesulfonic acid 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutyl ester (3f). Following the general procedure B, reaction of ethenesulfonate ester 1 (738 mg, 1.65 mmol) with pyrrolidine (382.8 mg, 5.4 mmol) in dichloromethane (50 cm3) afforded, after purification by medium pressure chromatography (20% acetone–hexane), the title compound (695 mg, 81%) as a light yellow oil (Found: C, 64.5; H, 8.5; N, 2.6. C28H43NO4SSi requires C, 64.95; H, 8.4; N, 2.7%); νmax (film)/cm−1 1354, 1165, 1106, 956, 822, 737 and 701; δH (300 MHz, CDCl3) 0.89 (6 H, s, (CH3)2C), 0.97 (9 H, s, (CH3)3C), 1.52 (2 H, t, J 6.7 Hz), 1.71–1.75 (4 H, br s), 2.48 (4 H, br s), 2.89 (2 H, dd, J 6.5, 9.0), 3.23 (2 H, dd, J 6.6, 9.0), 3.65 (2 H, t, J 6.7), 3.89 (2 H, s), 7.29–7.39 (6 H, m, Ph), 7.58–7.61 (4 H, m, Ph); δC (75.46 MHz, CDCl3) 19.5, 24.1, 24.6, 27.3, 34.2, 41.7, 50.0, 50.1, 54.1, 60.8, 78.0, 128.0, 130.0, 134.3, 135.9.
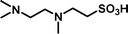
2-[(2-Dimethylaminoethyl)-methylamino]-ethanesulfonic acid 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutyl ester (3g). Following the general procedure B, reaction of ethenesulfonate ester 1 (716 mg, 1.60 mmol) with N,N,N′-trimethylethane-1,2-diamine (414 mg, 4.1 mmol) in dichloromethane (50 cm3) afforded, after purification by medium pressure chromatography (40% acetone–hexane + 5–7% Et3N), the title compound (717 mg, 81%) as a yellow oil (Found: C, 63.1; H, 8.8; N 4.9. C29H48N2O4SSi requires C, 63.5; H 8.8; N 5.1%); νmax (film)/cm−1 1352, 1165, 1103, 956, 822, 738 and 701; δH (300 MHz, CDCl3) 0.89 (6 H, s, (CH3)2C), 0.97 (9 H, s, (CH3)3C), 1.52 (2 H, t, J 6.7), 2.21 (9 H, s, NCH3), 2.26–2.50 (7 H, m), 2.85 (2 H, dd, J 6.2, 8.8), 3.18 (2 H, dd, J 6.2, 8.8), 3.65 (2 H, t, J 6.7), 3.89 (2 H, s), 7.29–7.39 (6 H, m, Ph), 7.58–7.61 (4 H, m, Ph); δC (75.46 MHz, CDCl3) 19.5, 24.6, 27.3, 34.2, 41.6, 42.5, 46.0, 48.5, 51.9, 55.7, 57.9, 60.8, 77.9, 128.0, 129.9, 134.3, 135.9.
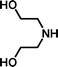
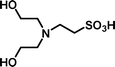
2-[Bis-(2-hydroxyethyl)-amino]-ethanesulfonic acid 4-(tert-butyldiphenylsilanyloxy)-2,2-dimethylbutyl ester (3h). Following the general procedure B, reaction of ethenesulfonate ester 1 (738 mg, 1.65 mmol) with diethanolamine (452 mg, 4.3 mmol) in dichloromethane (50 cm3) afforded, after purification by medium pressure chromatography (30% acetone–hexane), the title compound (629 mg, 69%) as an oil (Found: C, 60.2; H, 8.2; N, 2.5. C28H45NO6SSi requires C, 60.9; H, 8.2; N, 2.5%); νmax (film)/cm−1 1348, 1163, 1103, 951, 822, 738 and 702; δH (300 MHz, CDCl3) 0.89 (6 H, s, (CH3)2C), 0.97 (9 H, s, (CH3)3C), 1.52 (2 H, t, J 6.7), 2.59 (4 H, t, J 6), 3.00 (2 H, t, J 6.1), 3.16 (2 H, t, J 6.1), 3.56 (4 H, m), 3.66 (2 H, t, J 6.7), 3.91 (2 H, s), 7.29–7.39 (6 H, m, Ph), 7.57–7.61 (4 H, m, Ph); δC (125 MHz, CDCl3) 19.0, 24.1, 26.8, 33.7, 40.8, 48.2, 48.5, 56.5, 59.4, 60.3, 77.9, 127.7, 129.8, 133.5, 135.5.
2-Morpholin-4-yl-ethanesulfonic acid (4a). Following the general procedure C, reaction of sulfonate ester 3a (248 mg, 0.47 mmol) in THF (10 cm3) with TBAF (1 M in THF, 464 mg, 0.51 mmol) in THF (10 cm3) afforded a mixture of 4a and tetrabutylammonium species. Purification by ion exchange chromatography gave the title compound (66–72%) as a white solid, mp 277–280 °C (decomp.) (Found: C, 35.6; H 7.0; N 7.1. C6H13NO4S + ⅓ H2O requires C, 35.8; H, 6.9; N, 7.0%); νmax (film)/cm−1 1458, 1262, 1219, 1169, 1121, 1080, 1032, 978, 870, 822, 793 and 762; δH (300 MHz, D2O) 3.23–3.48 (8 H, m), 3.67–3.86 (4 H, br s); δC (125 MHz, D2O) 45.45, 52.88, 53.27, 64.62.
4-(2-Sulfoethyl)-piperazine-1-carboxylic acid tert-butyl ester (4b). Following the general procedure C, the reaction of sulfonate ester 3b (261 mg, 0.41 mmol) in THF (10 cm3) with TBAF (1 M in THF, 413 mg, 0.46 mmol) in THF (10 cm3) afforded a mixture of 4b and tetrabutylammonium species. Purification by ion exchange chromatography gave the title compound (91 mg, 75%) as a white solid, mp 228–229 °C (Found: C, 43.9; H, 7.5; N, 9.4. C11H22N2O5S + 0.5 H2O requires C, 43.6; H, 7.6; N, 9.2%); νmax (film)/cm−1 1693, 1406, 1365, 1227, 1158, 1070, 1036, 1008, 964, 934, 862, 802 and 756; δH (300 MHz, D2O) 1.35 (9 H, s), 3.26–3.51 (12 H, m); δC (75.48 MHz, D2O) 28.4, 41.4, 45.7, 52.7, 53.2, 83.7, 156.3.
2-(4-Acetylpiperazin-1-yl)-ethanesulfonic acid (4c). Following the general procedure C, reaction of sulfonate ester 3c (229 mg, 0.4 mmol) in THF (10 cm3) with TBAF (1 M in THF, 399 mg, 0.44 mmol) in THF (10 cm3) afforded a mixture of 4c and tetrabutylammonium species. Purification by ion exchange chromatography gave the title compound (77 mg, 82%) as a pink solid, mp 291–293 °C (decomp.) (Found: C, 39.6; H, 6.8; N, 11.7. C8H16N2O4S + ⅓ H2O requires C, 39.7; H, 6.9; N, 11.6%); νmax (film)/cm−1 1629, 1426, 1277, 1216, 1153, 1080, 1049, 997, 962, 838, 804, 764 and 721; δH (300 MHz, D2O) 2.06 (3 H, s, CH3), 3.27–3.48 (6 H, m), 3.50 (2 H, m), 3.79 (2 H, br s); δC (125 MHz, D2O) 21.0, 39.4, 44.0, 45.7, 52.4, 53.0, 173.5.
2-[4-(2-Methoxyethyl)-piperazin-1-yl]-ethanesulfonic acid (4d). Following the general procedure C, reaction of sulfonate ester 3d (282.6 mg, 0.478 mmol) in THF (10 cm3) with TBAF (1 M in THF, 475 mg, 0.53 mmol) in THF (10 cm3) afforded a mixture of 4d and tetrabutylammonium species. Purification by ion exchange chromatography gave the title compound (110 mg, 91%) as a pink solid, mp 210–212 °C (Found: C, 41.9; H, 7.9; N, 10.8. C9H20N2O4S + ⅓ H2O requires C, 41.8; H 8.1; N 10.8%); νmax (film)/cm−1 1332, 1267, 1225, 1165, 1118, 1078, 1040, 965 and 760; δH(300 MHz, D2O) 2.92–3.17 (8 H, m), 3.17–3.20 (6 H, m), 3.27 (3 H, s, CH3), 3.5–3.65 (2 H, m); δC(125 MHz, D2O) 47.9, 50.13, 51.94, 52.64, 56.5, 59.2, 66.55.
2-[4-(2-Hydroxyethyl)-piperidin-1-yl]-ethanesulfonic acid (4e). Following the general procedure C, reaction of sulfonate ester 3e (274 mg, 0.476 mmol) in THF (10 cm3) with TBAF (1 M in THF, 469 mg, 0.52 mmol) in THF (10 cm3) afforded a mixture of 4e and tetrabutylammonium species. Purification by ion exchange chromatography gave the title compound (91 mg, 81%) as a beige solid, mp 249–251 °C (Found: C, 44.7; H, 8.0; N, 5.9. C9H19NO4S + 0.25 H2O requires C, 44.7; H, 8.1; N, 5.8%); νmax (film)/cm−1 3372, 1451, 1221, 1164, 1038, 962, 941, 916, 816 and 758; δH(300 MHz, D2O) 1.39–1.46 (4 H, m), 1.89–1.94 (2 H, m), 2.90 (1 H, dt, J 2.4, J 12.9), 3.19–3.56 (9H, m); δC (125 MHz, D2O) 30.0, 30.5, 37.9, 45.9, 53.0, 54.2, 59.7.
2-Pyrrolidin-1-yl-ethanesulfonic acid (4f). Following the general procedure C, reaction of sulfonate ester 3f (244 mg, 0.471 mmol) in THF (10 cm3) with TBAF (1 M in THF, 469 mg, 0.52 mmol) in THF (10 cm3) afforded a mixture of 4f and tetrabutylammonium species. Purification by ion exchange chromatography gave the title compound (66 mg, 78%) as a white solid, mp 216–218 °C (Found: C, 38.6; H, 7.1; N, 7.5. C6H13NO3S + 0.5 H2O requires C, 38.3; H, 7.5; N, 7.4%); νmax (film)/cm−1 1455, 1215, 1163, 1032 and 911; δH (300 MHz, D2O) 1.84–2.08 (4 H, m), 2.98–3.06 (2 H, m), 3.18 (2 H, t, J 7.3), 3.44 (2 H, t, J 7.3), 3.56–3.64 (2 H, m); δC (125 MHz, D2O) 23.54, 47.40, 51.22, 55.47.
2-[(2-Dimethylaminoethyl)methylamino]-ethanesulfonic acid (4g). Following the general procedure C, reaction of sulfonate ester 3g (255 mg, 0.46 mmol) in THF (10 cm3) with TBAF (1 M in THF, 462 mg, 0.51 mmol) in THF (10 cm3) afforded a mixture of 4g and tetrabutylammonium species. Purification by ion exchange chromatography gave the title compound (90 mg, 92%) as a yellow oil (Found: C, 36.9; H, 8.8; N, 11.8. C7H18N2O3S + H2O requires C, 36.8; H, 8.8; N, 12.3%); νmax (film)/cm−1 1647, 1467, 1175, 1034 and 731; δH (300 MHz, D2O) 2.31 (3 H, s), 2.80 (6 H, s), 2.82–2.92 (4 H, m), 3.02–3.07 (2 H, m), 3.22 (2 H, t, J 6.6); δC (75.48 MHz, D2O) 41.87, 44.06, 48.7, 51.6, 52.8, 55.5.
2-[Bis-(2-hydroxyethyl)-amino]-ethanesulfonic acid (4h). Following the general procedure C, reaction of sulfonate ester 3h (260 mg, 0.472 mmol) in THF (10 cm3) with TBAF (1 M in THF, 469 mg, 0.52 mmol) in THF (10 cm3) afforded a mixture of 4h and tetrabutylammonium species. Purification by ion exchange chromatography give the title compound (91 mg, 90%) as a white solid, mp 159–160 °C (Found: C, 32.9; H, 7.0; N, 6.4. C6H15NO5S + 0.5 H2O requires C, 32.4; H, 7.3; N, 6.3%); νmax (film)/cm−1 3389, 3044, 1327, 1158, 1088, 1032, 961, 891, 862, 758 and 735; δH (300 MHz, D2O) 3.27 (2 H, t, J 7.0), 3.33–3.37 (4 H, m), 3.62 (2 H, t, J 6.9), 3.81–3.85 (4 H, m); δC (125 MHz, D2O) 45.41, 50.90, 55.95, 56.32.
Acknowledgements
We thank Cancer Research UK, MRC and OSI Pharmaceuticals for support of this research.References
- M. E. Brady, D. M. Ozanne, L. Gaughan, I. Waite and S. Cook, J. Biol. Chem., 1999, 274, 17599 CrossRef CAS
 ; L. Gaughan, M. E. Brady, S. Cook, D. E. Neal and C. N. Robson, J. Biol. Chem., 2001, 276, 46841 CrossRef CAS ; L. Gaughan, I. R. Logan, S. Cook, D. E. Neal and C. N. Robson, J. Biol. Chem., 2002, 277, 25904 CrossRef CAS ; K. Halkidou, V. J. Gnanapragasam, P. Mehta, I. R. Logan, M. E. Brady, S. Cook, H. Y. Leung, D. E. Neal and C. N. Robson, Oncogene, 2003, 22, 2466 CrossRef CAS .
; L. Gaughan, M. E. Brady, S. Cook, D. E. Neal and C. N. Robson, J. Biol. Chem., 2001, 276, 46841 CrossRef CAS ; L. Gaughan, I. R. Logan, S. Cook, D. E. Neal and C. N. Robson, J. Biol. Chem., 2002, 277, 25904 CrossRef CAS ; K. Halkidou, V. J. Gnanapragasam, P. Mehta, I. R. Logan, M. E. Brady, S. Cook, H. Y. Leung, D. E. Neal and C. N. Robson, Oncogene, 2003, 22, 2466 CrossRef CAS . - D. E. Metzler, in Biochemistry, Academic Press, London, 2nd edn, 2003, vol. 2, p. 1407 Search PubMed .
- L. Bischoff, C. David, B. Roques and M. Fournie-Zaluski, J. Org. Chem., 1999, 64, 1420 CrossRef CAS ; C. David, L. Bischoff, H. Meudal, A. Mothe, N. De Mota, S. DaNascimento, C. Llorens-Cortes, M. Fournie-Zaluski and B. Roques, J. Med. Chem., 1999, 42, 5197 CrossRef CAS .
- L. Yan and C. E. Müller, J. Med. Chem., 2004, 47, 1031 CrossRef CAS .
- J. Wrobel, J. Rogers, D. Green and W. L. Kao, Synth. Commun., 2002, 32, 2695 CrossRef CAS .
- B. Musicki and T. S. Widlanski, J. Org. Chem., 1990, 55, 4231 CrossRef CAS ; B. Musicki and T. S. Widlanski, Tetrahedron Lett., 1991, 32, 1267 CrossRef CAS .
- M. Xie and T. S. Widlanski, Tetrahedron Lett., 1996, 37, 4443 CrossRef CAS .
-
(a) W. E. Truce and D. J. Vrencur, J. Org. Chem., 1970, 35, 1226 CrossRef CAS ;
(b) J. C. Roberts, H. Gao, A. Gopalsamy, A. Kongsjahju and R. J. Patch, Tetrahedron Lett., 1997, 38, 355 CrossRef CAS .
- B. G. Avitabile, C. A. Smith and D. B. Judd, Org. Lett., 2005, 7, 843 CrossRef CAS .
- A. Hari and B. Miller, Org. Lett., 1999, 1, 2109 CrossRef CAS .
- D. Klamann and G. Hofbauer, Chem. Ber., 1953, 86, 1246 CrossRef CAS ; J. E. Richman and T. J. Atkins, J. Am. Chem. Soc., 1997, 96, 2268 .
- A. K. Andrianov, A. Marin, J. Chen, J. Sargent and N. Corbett, Macromolecules, 2004, 37, 4075 CrossRef CAS .
- P. Klan, A. P. Pelliccioli, T. Pospisil and J. Wirz, Photochem. Photobiol. Sci., 2002, 1, 920 RSC .
- G. W. Kenner, J. R. McDermott and R. C. Sheppard, J. Chem. Soc. D, 1971, 636 RSC .
- For reviews see:
(a) F. Guillier, D. Orain and M. Bradley, Chem. Rev., 2000, 100, 2091 CrossRef ;
(b) P. Heidler and A. Link, Bioorg. Med. Chem., 2005, 13, 585 CrossRef CAS .
- J. F. King, S. M. Loosmore, M. Aslam, J. D. Lock and M. J. McGarrity, J. Am. Chem. Soc., 1982, 104, 7108 CrossRef CAS .
- Preparation of the TIPS-derivative: F. Richter, M. Bauer, C. Perez, C. Maichle-Moessmer and M. E. Maier, J. Org. Chem., 2002, 67, 2474 Search PubMed .
- A. Le Berre, A. Étienne and B. Dumaitre, Bull. Soc. Chim. Fr., 1970, 3, 946 CAS .
- R. M. Beesley, C. K. Ingold and J. F. Thorpe, J. Chem. Soc., Trans., 1915, 107, 1080 RSC ; C. K. Ingold, S. Saka and J. F. Thorpe, J. Chem. Soc., Trans., 1922, 121, 1177 RSC .
- M. Mazet and M. Desmaison-Brut, Bull. Soc. Chim. Fr., 1971, 2656 CAS .
- Y.-L. Chou, M. M. Morrisey and R. Mohan, Tetrahedron Lett., 1998, 39, 757 CrossRef CAS .
- L. S. Simpson and T. S. Widlanski, J. Am. Chem. Soc., 2005, 128, 1605 .
|
| This journal is © The Royal Society of Chemistry 2007 |
Click here to see how this site uses Cookies. View our privacy policy here.