CIAT with simultaneous epimerization at two stereocenters. Synthesis of substituted β-methyl-α-homophenylalanines†
Dušan
Berkeš
*a,
Pavol
Jakubec
a,
Dagmar
Winklerová
a,
František
Považanec
a and
Adam
Daich
*b
aDepartment of organic chemistry, Slovak University of Technology, Radlinského 9, SK-81237, Bratislava, Slovakia. E-mail: dusan.berkes@stuba.sk; Fax: +421 2 52968560; Tel: +421 2 52968560
bLaboratoire de Chimie, URCOM, UFR des Techniques de l'Université du Havre, 25, rue Philippe Lebon, B.P.: 540, F-76058, Le Havre Cedex, France. E-mail: adam.daich@univ-lehavre.fr; Fax: +33 02-32-74-43-91; Tel: +33 02-32-74-44-03
First published on 23rd November 2006
Abstract
Diastereoselective aza-Michael additions of phenylethylamine to 3-aroylbutenoic acids are reported. During these processes, efficient control over two new stereogenic centers on the Michael acceptor has been possible via crystallization-induced asymmetric transformation (CIAT). As an application, a convenient two-step synthesis of anti-β-methylhomophenylalanines is also described.
Introduction
Crystallization-induced asymmetric transformation (CIAT) of diastereomers or crystallization-induced diastereomer transformation1 is an efficient tool for stereoselective synthesis based on thermodynamic control. Because it is not necessary to work at low temperature and readily available chiral auxiliaries or chirality mediators can be used to build a new stereogenic center, CIAT is an effective means to develop highly diastereoselective processes especially on an industrial scale.2 In spite of the exceptional effectiveness of CIAT processes the number of known applications is relatively low. The critical point in the CIAT development is the compatibility of the epimerization (racemization) conditions in the solution with the conditions for the crystal growth and nucleation.3,4 More than half of the papers on racemization deal with amino acids or their derivatives.5γ-Oxo substituted derivatives of amino acids are derivatives with high potential for further development, and their synthesis has accordingly been lately studied in considerable detail. The principal strategies thereby pursued were as follows: catalytic, enantioselective Mannich-type reactions,6,7 “chiral pool” syntheses from available amino acids, mainly of the L-series,8–10 and alternatively using the tandem reaction sequence, consisting of aza-Michael addition of chiral N-nucleophiles to aroylacrylic acids, followed by crystallization-induced asymmetric transformation (CIAT). The latter route, owing to its technological robustness and simplicity of operation, was successfully used for accessing enantiomerically pure precursors of ACE inhibitors.11,12
Our research program is focused on applications of CIAT to the synthesis of γ-oxo and γ-hydroxy substituted α-amino acids. The success of such transformations is based on the formation of amino acids only slightly soluble at their isoelectric point and on reversible aza-Michael addition, the retro-Michael being catalyzed by excess of the base.13–16 Recently we have described a remarkable phenomenon, whereby simultaneous epimerization occurred at two stereogenic centers. This resulted in the formation of only one of the four possible stereoisomers in high yield, as well as excellent diastereomeric and enantiomeric purity.17 Such CIAT applications are very rare in the literature.18,19
Here we would like to present an interesting application of this concept to the synthesis of anti-β-methyl-α-homophenylalanines and their γ-hydroxy derivatives with high diastereoselectivity control over two new stereogenic centers. β-Methyl-α-homophenylalanine forms a vital part of cytotoxic depsipeptide kulokekahilide-1.20 Interestingly, the same constrained amino acid (4-phenylvaline) can be found in the potential anticancer agent dolastatin-16 and also in the cyclic depsipeptide homodolastatin-16.21,22 The stereochemistry of 2-amino-3-methyl-4-phenylbutanoic acid in the last two natural species remains unassigned. In addition, γ-hydroxy substituted β-methyl-α-homophenylalanines can be found in many biologically active substances like antifungal nikkomycins23 and immunosuppresive cymbimycins.24
Results and discussion
The starting unsaturated acids 2a–c have been prepared by modified acid catalyzed condensation of the corresponding propiophenones 1a–c with glyoxylic acid (Scheme 1).25 The Friedel–Crafts reaction of aromatics with citraconic anhydride exhibits low regioselectivity and stereoselectivity and the formation of the (Z)-stereomer in the form of cyclic tautomeric lactone was observed.26 In our optimized conditions the desired (E)-stereomers 2a–c were prepared in yields ranging from 51 up to 73% and in E : Z ratio more than 81 : 19, which can be increased up to 96 : 4 by one crystallization. 4-Methoxy substituted propiophenones 2b,c were prepared by highly regioselective LiClO4-catalyzed acylation.27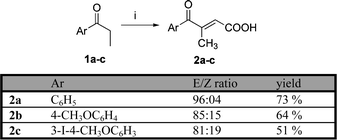 | ||
| Scheme 1 (i) HOOC-CHO·H2O, H2SO4, dioxane, reflux, 1.5 h. | ||
Two new stereogenic centres are formed by conjugated addition of amines on Michael acceptors 2a–c. For the design of a successful CIAT process the conditions for an effective equilibrium between all the possible stereomers in solution and the phase equilibria in heterogeneous mixture between the precipitated solid and solution have to be precisely ascertained.
Firstly, the conditions for the addition of benzylamine to unsaturated acid 2a were optimized. Stirring the starting 0.21 M aqueous solution of 2a with benzylamine (1.1 equiv.) at 40 °C caused slow precipitation of both adducts and an effective epimerization between them in alkaline solution (Fig. 1). After several days the filtration of the reaction suspension allows one to obtain only one of two possible diastereomers in high yield and with excellent diastereomeric purity (anti-3a; 70%; dr 97 : 3). As can be gleaned from Fig. 1A), the ratio of the diastereomeric adducts at the initial stages is the opposite (anti : syn = 17 : 83 after 2 h). A similar reaction course (with small alteration of the solvent concentration) has been observed for the unsaturated acids 2b,c (Scheme 2).
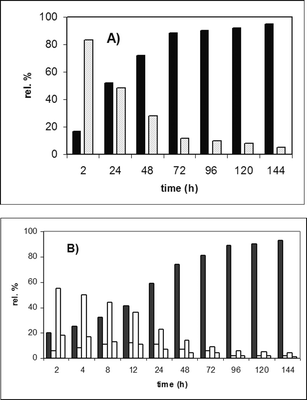 | ||
Fig. 1 Diastereomer distribution (HPLC experiments) A) 0.21 M of 2a in water with benzylamine (1.1 equiv.) at 40 °C ■
3a, ![[square tinted (light)]](https://www.rsc.org/images/entities/char_e068.gif) syn-diastereomer; B) 0.26 M of 2b in water with (S)-PEA (1.1 equiv.) at 40 °C ■
4b, □ other diastereomers.
syn-diastereomer; B) 0.26 M of 2b in water with (S)-PEA (1.1 equiv.) at 40 °C ■
4b, □ other diastereomers. | ||
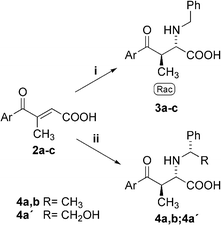 | ||
| Scheme 2 aza-Michael addition of amine and CIAT process. (i) 1.1 equiv. of BnNH2, water, 40 °C, 7 days, filtration; (ii) 4a,b: 1.1 equiv. of (S)-PEA, water, 40 °C, 7 days, filtration; 4a: 1.1 equiv. of (R)-phenylglycinol, CH2Cl2, 25 °C, 14 days, filtration. | ||
Having in hand the optimized process for the benzylamine addition the reactions of chiral (S)-phenylethylamine ((S)-PEA) and (R)-phenylglycinol in tandem with an effective CIAT process have been studied. The best results were obtained with (S)-PEA in conditions consistent with benzylamine addition (water, 0.26 M solution, 40 °C, 7 days). Only one of the four possible isomers has been separated from the reaction mixture by simple filtration (4a: 70% yield, dr 99 : 0 : 1 : 0; 4b: 60% yield, dr 99 : 0 : 1 : 0 in HPLC succession). In the case of (R)-phenylglycinol only dichloromethane has been found to be a suitable solvent for the CIAT process with 2a, however, the yield of precipitated product was low (4a′:18% after 14 days, dr 1 : 99 : 0 : 0).
The course of the CIAT process for the (S)-PEA addition on acid 2b is outlined on Fig. 1B). Also in this case at the initial stages of the transformation the kinetically favoured diastereomers predominated in the reaction mixture. This is in agreement with the benzylamine addition. However as the CIAT progressed in time 4b clearly became a major product with concomitant decline of the content of all other isomers in the reaction suspension.
The prepared γ-oxo-α-amino acids 3a–c or 4a,b are stable as solids, however they decompose slowly especially in alkaline solution. The reduction of the carbonyl group has been found as a tool for the stabilization of the newly formed stereogenic centers.28
A stereodivergent route to both diastereomeric γ-hydroxy-α-amino acids 5a,b and 6a,b was developed (Scheme 3) using sodium borohydride in convenient reaction conditions. The application of sodium borohydride in methanol produces the 3,4-syn-isomers 5a,b in high yield and excellent diastereomeric purity via 1,2-stereoinduction and it is in agreement with the known results on related systems.29 The 3,4-anti-isomers 6a,b were prepared using our previously reported catalytic reduction with NaBH4–cat. MnCl2·4H2O system in methanol by 1,3-asymmetric induction via Mn2+ chelation.28 The chemoselective catalytic N-debenzylation of 5a,b and 6a,b produces the 2-amino-4-aryl-4-hydroxy-3-methylbutanoic acids 7a,b and 8a,b, the non-natural analogues of N-terminal amino acid of nikkomycins.
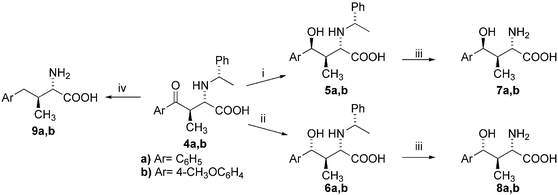 | ||
| Scheme 3 Stereodivergent reduction and catalytic hydrogenation of oxoamino acids 4. (i) NaBH4, MeOH, 0–5 °C, 5 : 6 > 92 : 8; (ii) NaBH4, MnCl2, MeOH, 0–5 °C, 5 : 6 > 5 : 95; (iii) 1 equiv. HBr, H2/Pd–C, MeOH–H2O, 25 °C; (iv) 3 equiv. HBr, H2/Pd–C, MeOH–H2O, 40 °C, 24 h. | ||
Elucidation of the relative and absolute configuration
Determination of the relative configuration on the newly formed stereogenic centres was based on the results obtained from NOE experiments, which were performed on cyclic derivatives 10a,b and 11a,b.Acid-catalyzed lactonization of the phenyl substituted hydroxyamino acids 5a,6a under simple stirring in diluted HCl led to the corresponding crystalline lactones 10a,11a in high yield. One recrystallization seems to be sufficient to obtain these nicely crystalline hydrochlorides in excellent stereochemical homogeneity (dr > 99 : 1). In the case of methoxy substituted derivatives 5b,6b the lactonization took place smoothly under mild conditions (3 M HCl, 4 h, rt), however, in both cases the all cis-isomer 10b was isolated in excellent both yield and purity. Such a result accords with our recently described CIAT application on closely related derivatives.16
Gratifyingly, the employment of DCC in our previously described conditions allowed us to prepare the desired lactone 11b under mild conditions (Scheme 4). As expected, the NOE's observed between protons at C-2, C-3 and C-4 testified to the relative all cis-configuration of 10b (Scheme 4). Similarly the NOE data clearly confirmed the expected 3,4-trans-relative configuration on the lactone 11b. The relative configurations of all hydroxyamino acids 5,6 and corresponding lactones 10a,11a were tentatively assigned on the basis of these results.
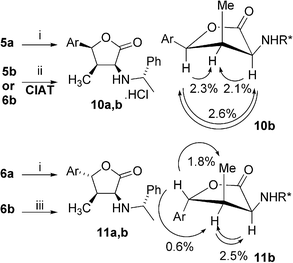 | ||
| Scheme 4 Lactone formation, establishment of relative configuration. (i) 10a: 3 M HCl, rt, 24 h, filtration, 10 : 11 ratio 98 : 2, 90%; 11a: 8 M HCl, 40 °C, 20 h, filtration, 10 : 11 ratio 3 : 97, 76%; (ii) 10b: 3 M HCl, rt, 4 h, filtration, 10 : 11 ratio 95 : 5, 89–90%; (iii) 11b: DCC, CH2Cl2, rt, 20 h, chromatography, 10 : 11 ratio 2 : 98, 50%. | ||
Absolute configuration assignment of the carbons C-2 and C-3 of the parent adducts 4a,b and 4a′ was made by their transformation to intermediates of biologically important compounds and at the same time the relative configuration was also tested. Pd-catalyzed hydrogenation proceeds smoothly in an EtOH–H2O combination with excess of HBr. No epimerization to the (2S,3S)-2-amino-3-methyl-4-phenylbutanoic acid 9a has been observed. The NMR spectral data and optical properties are in agreement with those published by Kimura et al. in the kulokekahelide studies.20
Furthermore, the methoxy substituted hydroxyamino acids 7b,8b were transferred to the known Boc-protected lactones 12b,13b30 (Scheme 5)—intermediates of the nikkomycin-B synthesis—and their NMR spectral data as well as their specific optical rotation confirm the (2S,3R,4S)-12b or (2S,3R,4R)-13b configuration and conclude the absolute and relative structural assignment of methoxy substituted derivatives given above.
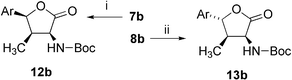 | ||
| Scheme 5 Lactone 12b,13b synthesis, absolute configuration elucidation. (i) a: 12 M HCl, rt, 1 h, filtration, 100%; b: (Boc)2O, NEt3, dioxane, 50%, 12 : 13 ratio 98 : 2; (ii) a: Boc2O, NEt3, CH2Cl2, 30 °C, b: DCC, CH2Cl2, 5 min (dr 81 : 19), chromatography, 28% of 13b, 12 : 13 ratio 2 : 98. | ||
Conclusions
We have successfully broadened the scope of CIAT in the conjugate addition of N-nucleophiles to substituted aroylacrylic acids. We have demonstrated an efficient CIAT methodology, enabling an efficient stereocontrol over two new stereogenic centers. We report herein a two-step and inexpensive preparation of the 4-aryl substituted 2-amino-3-methylbutanoic acids and their 4-hydroxy substituted derivatives, respectively, with high degree of both diastereomeric and enantiomeric purity.Acknowledgements
Financial support by the Slovak Grant Agency No. 1/2469/05 and NMR measurements provided by the Slovak State Programme Project No. 2003SP200280203 are gratefully acknowledged.References
- K. M. J. Brands and A. J. Davies, Chem. Rev., 2006, 106, 2711–2733 CrossRef CAS.
- N. G. Anderson, Org. Process Res. Dev., 2005, 9, 800–813 Search PubMed.
- J. W. Schroer and K. M. Ng, Ind. Eng. Chem. Res., 2003, 42, 2230–2244 CrossRef CAS.
- K. Kinbara, Synlett, 2005, 732–743 CrossRef CAS.
- E. J. Ebbers, G. J. A. Ariaans, J. P. M. Houbiers, A. Bruggink and B. Zwanenburg, Tetrahedron, 1997, 53, 9417–9476 CrossRef CAS.
- D. Ferraris, B. Young, C. Cox, T. Dudding, W. J. Drury, lll, L. Ryzhkov, A. E. Taggi and T. Lectka, J. Am. Chem. Soc., 2002, 124, 67–77 CrossRef CAS.
- S. Kobayashi, R. Matsubara, Y. Nakamura, H. Kitagawa and M. Sugiura, J. Am. Chem. Soc., 2003, 125, 2507–2515 CrossRef CAS.
- A. S. Golubev, N. Sewald and K. Burger, Tetrahedron, 1996, 52, 14757–14776 CrossRef.
- R. F. W. Jackson, R. J. Moore, C. S. Dexter, J. Elliott and C. E. Mowbray, J. Org. Chem., 1998, 63, 7875–7884 CrossRef CAS.
- I. Rilatt, L. Caggiano and R. F. W. Jackson, Synlett, 2005, 2701–2719 CAS.
- M. Yamada, N. Nagashima, J. Hasegawa and S. Takahashi, Tetrahedron Lett., 1998, 39, 9019–9022 CrossRef CAS.
- H. Urbach and R. Henning, Tetrahedron Lett., 1984, 25, 1143–1146 CrossRef CAS.
- A. Kolarovič, D. Berkeš, P. Baran and F. Považanec, Tetrahedron Lett., 2001, 42, 2579–2582 CrossRef CAS.
- P. Jakubec, D. Berkeš and F. Považanec, Tetrahedron Lett., 2004, 45, 4755–4758 CrossRef CAS.
- D. Berkeš, A. Kolarovič, R. G. Raptis and P. Baran, J. Mol. Struct., 2004, 697, 101–107 CrossRef CAS.
- D. Berkeš, A. Kolarovič, R. Manduch, P. Baran and F. Povaanec, Tetrahedron: Asymmetry, 2005, 16, 1927–1934 CrossRef CAS.
- A. Kolarovič, D. Berkeš, P. Baran and F. Povaanec, Tetrahedron Lett., 2005, 46, 975–978 CrossRef CAS.
- H. T. Openshaw and N. Whittaker, J. Chem. Soc., 1963, 1461–1471 RSC.
- R. D. Clark, J. R. Kern, L. J. Kurz and J. T. Nelson, Heterocycles, 1990, 31, 353–366 CrossRef CAS.
- J. Kimura, Y. Takada, T. Inayoshi, Y. Nakao, G. Goetz, W. Y. Yoshida and P. J. Scheuer, J. Org. Chem., 2002, 67, 1760–1767 CrossRef CAS.
- L. M. Nogle and W. H. Gerwick, J. Nat. Prod., 2002, 65, 21–24 CrossRef CAS.
- M. T. Davies-Coleman, T. M. Dzeha, C. A. Gray, S. Hess, L. K. Pannell, D. T. Hendricks and C. A. Arendse, J. Nat. Prod., 2003, 66, 712–715 CrossRef CAS.
- W. A. König, H. Hahn, R. Rathmann, W. Hass, A. Keckeisen, H. Hagenmaier, C. Bormann, W. Dehler, R. Kurth and H. Zähner, Liebigs Ann. Chem., 1986, 407–421 CrossRef.
- T. Fehr, V. F. J. Quesniaux, J. J. Sanglier, L. Oberer, L. Gschwind, M. Ponelle, W. Schilling, S. Wehrli, A. Enz, G. Zenke and W. Schuler, J. Antibiot., 1997, 893–899 CAS.
- F. J. McEvoy and G. R. Allen, J. Org. Chem., 1973, 38, 4044–4048 CrossRef CAS.
- M. Kuchař, V. Vosátka, M. Poppová, E. Knězová, V. Ponajotová, H. Tomková and J. Taimr, Collect. Czech. Chem. Commun., 1995, 1026–1033 CrossRef CAS.
- G. Bartoli, M. Bosco, E. Marcantoni, M. Massaccesi, S. Rinaldi and L. Sambri, Tetrahedron Lett., 2002, 43, 6331–6333 CrossRef CAS.
- D. Berkeš, A. Kolarovič and F. Považanec, Tetrahedron Lett., 2000, 41, 5227–5260.
- S. Kobayashi, T. Hamada and K. Manabe, J. Am. Chem. Soc., 2002, 124, 5640–5641 CrossRef CAS.
- J. Barluenga, A. L. Viado, E. Aquilar, S. Fustero and B. Olano, J. Org. Chem., 1993, 58, 5972–5975 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H NMR and 13C NMR spectra are provided for all the products reported herein. See DOI: 10.1039/b613103d |
| This journal is © The Royal Society of Chemistry 2007 |