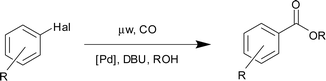
Alkoxycarbonylation of aryl iodides using gaseous carbon monoxide and pre-pressurized reaction vessels in conjunction with microwave heating†
Chad M.
Kormos
and
Nicholas E.
Leadbeater
*
Department of Chemistry, University of Connecticut, 55 North Eagleville Rd, Storrs, CT 06269-3060, USA. E-mail: nicholas.leadbeater@uconn.edu; Fax: +1 860 486 2981; Tel: +1 860 486 5076
First published on 14th November 2006
Abstract
The microwave-promoted alkoxycarbonylation of aryl iodides using reaction vessels pre-pressurized with carbon monoxide is reported. Reactions are performed using 0.1 mol% palladium acetate as catalyst, DBU as base and are complete within 20–30 min. A range of aryl iodide substrates can be converted to the corresponding esters using this methodology. Primary and secondary alcohols work well whereas a tertiary alcohol substrate proves less reactive. The potential for scale-up of the reaction has also been explored.
Introduction
The palladium catalyzed carbonylation of aryl halides is a reaction of both academic and industrial relevance since a range of products including amides, esters and carboxylic acids can be prepared in one step.1–4 A range of palladium complexes have been prepared and screened for activity in these reactions, the majority bearing highly basic monodentate or bidentate phosphine ligands. These complexes are used in an attempt to limit clustering and agglomeration of palladium atoms in the presence of CO to give catalytically inactive species. Issues associated with the use of phosphines are that they are often difficult to prepare and can be highly air sensitive. There have been some recent reports of stable phosphine-containing palladium complexes for use in carbonylation chemistry.5 These go some way to alleviating some of the issues of using phosphines and also expand the substrate scope of the reaction. However simpler, cheaper and more easy to use catalyst systems would also be beneficial.In our laboratory we have been focusing attention on the use of simple ligandless palladium complexes as catalysts for carbonylation chemistry. We perform our reactions using microwave heating, this being a valuable tool for synthetic chemists because it is possible to enhance the rate of reactions and, in many cases, improve product yields.6,7 Most scientific microwave systems do not have a commercially available gas-loading accessory and the glass tubes used for reactions have a pressure limit of 20–30 bar. As a result, with the combination of a pressure of reactive gas and the autogenic pressure of solvents at elevated temperatures, there is a limit to the temperature to which reaction mixtures can be heated. These factors have been reflected in the scarcity of reports of organic synthesis in pre-pressurized vessels using microwave heating.8,9 Using a newly developed dedicated multi-mode microwave reactor we have shown that it is possible to perform hydroxycarbonylation reactions using aryl iodides as substrates.10 The chemistry is carried out in heavy-walled quartz reaction vessels with operating limits of 80 bar. The reactor is also equipped with a gas-loading interface, allowing the vessels to be pre-pressurized to up to 20 bar prior to placing in the microwave cavity.11,12 We performed our hydroxycarbonylation reactions using palladium acetate as the catalyst and water as the solvent and a CO pressure of 10 bar. The reactions were complete within 20 min of microwave heating and no additional ligand is required. A range of aryl iodides were converted to the benzoic acids. Strategies for performing carbonylation chemistry using microwave heating but without the need for using gaseous carbon monoxide have also been reported. Larhed and co-workers have used Mo(CO)6 as a source of carbon monoxide for the preparation of amides, esters and carboxylic acids from aryl halides.13 Advantages of using Mo(CO)6 as a replacement for gaseous CO include the fact that it is a solid and is easily used on a small scale with commercially available monomode microwave apparatus with no modification required. However, Mo(CO)6 is toxic and its use results in metal waste; this being a particular problem if the reaction is to be scaled up. We therefore wanted to expand our carbonylation chemistry to the preparation of esters. Our results are presented here.
Results and discussion
With ethanol as a substrate and solvent, our starting point was to use reaction conditions similar to those developed for the hydroxycarbonylation chemistry in water; namely using ligandless palladium sources as catalysts and sodium carbonate as a base. Pre-loading reaction vessels to 10 bar with CO, we screened a range of conditions for the conversion of 4-iodoanisole to ethyl 4-methoxybenzoate. In the screening reactions, absolute (200 proof) ethanol was used. Key results are shown in Table 1. Using similar conditions to those employed in our hydroxycarbonylation reactions [0.1 mol% Pd(OAc)2, 1.1 eq. Na2CO3, 165 °C] we obtained only a 7% yield of the desired ester product (Table 1, entry 1). Reducing the reaction temperature from 165 °C to 125 °C gave a 16% yield of the desired ester (Table 1, entry 2). This could be improved to 45% by changing the base from Na2CO3 to Cs2CO3 (Table 1, entry 3). We next decided to screen an organic base in the reaction. The use of amines as bases for alkoxycarbonylation reactions has been reported before.14,15 Using 1.1 eq DBU (1,8-diazobicyclo[5.4.0]undec-7-ene) as the base, a 91% yield of the desired ester was obtained (Table 1, entry 4). Triethylamine, another reported organic base, gave a lower product yield (Table 1, entry 5). Having found a good base for the reaction, we next wanted to see if the temperature could be reduced further but moving from 125 °C to 115 °C had a slightly deleterious effect on the product yield (Table 1, entry 6). Working at 125 °C, we tried reducing the catalyst loading by a factor of 10 from 0.1 mol% to 0.01 mol%. This resulted in a significant drop in yield (Table 1, entry 7), even when the reaction temperature was increased to 135 °C (Table 1, entry 8). Thus our optimum conditions were: 0.1 mol% Pd(OAc)2 as catalyst, 1.1 eq. DBU as the base, heating to 125 °C and holding at this temperature until a total reaction time of 20 min had elapsed.|
|
||
|---|---|---|
| Entry | Reaction conditionsa,b | Yield (%) |
| a Reactions were run in a sealed tube, pre-loaded with 10 bar CO, using 1.5 mmol 4-iodoansole and 15 mL 200 proof ethanol. An initial microwave irradiation power of 1000 W was used, the temperature being ramped from r.t. to that shown and held until a total reaction time of 20 min had elapsed. b For clarity, changes in reaction conditions from entry 1 are noted in bold. | ||
| 1 | 0.1 mol% Pd(OAc)2, 1.1 eq. Na2CO3, 165 °C | 7 |
| 2 | 0.1 mol% Pd(OAc)2, 1.1 eq. Na2CO3, 125 °C | 16 |
| 3 | 0.1 mol% Pd(OAc)2, 1.1 eq. Cs2CO3, 125 °C | 45 |
| 4 | 0.1 mol% Pd(OAc)2, 1.1 eq. DBU, 125 °C | 91 |
| 5 | 0.1 mol% Pd(OAc)2, 1.1 eq. NEt3, 125 °C | 60 |
| 6 | 0.1 mol% Pd(OAc)2, 1.1 eq. DBU, 115 °C | 86 |
| 7 | 0.01 mol% Pd(OAc) 2 , 1.1 eq. DBU, 125 °C | 12 |
| 8 | 0.01 mol% Pd(OAc) 2 , 1.1 eq. DBU, 135 °C | 24 |

Of interest is that the conditions for alkoxycarbonylation are milder than those used for hydroxycarbonylation. In particular, the reaction temperature is 40 °C lower. We attribute the differences to the fact that carbon monoxide is significantly more soluble in ethanol than water.
Before continuing to screen a range of aryl halide substrates, we had to overcome a problem with the isolation of the ester product. Performing a simple aqueous/organic work-up of the reaction mixture left significant quantities of ethanol in the organic layer. Subsequent removal of the organic solvent and ethanol on a rotary evaporator resulted in significant loss of ester product. We believe that an azeotrope forms, the ethanol facilitating the evaporation of the product ester. To overcome this problem, we had to develop an alternative work-up procedure. An amount of brine equal to the volume of alcohol was added. The mixture was then shaken with diethyl ether (3 × 15 mL) and the organic washings combined. Addition of hexane to the combined organics, resulted in a biphasic solution. One layer comprised of the hexane and ether and contained the ester product. The other layer contained the unreacted ethanol, draining of which then allowed for the ester to be recovered without loss.
We screened a range of aryl halide substrates using our optimal reaction conditions. The results are shown in Table 2. As with our hydroxycarbonylation protocol, only aryl iodides can be converted to esters; aryl bromides are unreactive (Table 2, entry 3). A range of aryl iodides can be converted to the ethyl esters including ortho-substituted examples (Table 2, entries 9 and 10). A representative heteroaromatic substrate, 3-iodopyridine, gave a good yield of the desired ester (Table 2, entry 11). This is noteworthy since, in the case of the hydroxcarbonylation reaction, only low yields of the desired acid product were obtained when using heteroaromatic substrates. This is attributed in part to competitive decomposition as well as to the difficulty in isolating the acid product from the reaction mixture.
|
|
|||
|---|---|---|---|
| Entry | Aryl halide | Alcohol | Product yield (%) |
| a Reactions were run in a sealed tube, pre-loaded with 10 bar CO, using 1.0 mmol aryl halide, 10 mL of the desired alcohol, 0.1 mol% Pd(OAc), and 1.1 mmol DBU. An initial microwave irradiation power of 1000 W was used, the temperature being ramped from r.t. to 125 °C and held until a total reaction time of 20 min had elapsed. b Reaction time extended to 30 min. | |||
| 1 |

|
EtOH | 90 |
| 2 |
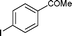
|
EtOH | 99 |
| 3 |

|
EtOH | 0 |
| 4 |

|
EtOH | 91 |
| 5 |
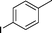
|
EtOH | 94 |
| 6 |

|
EtOH | 91 |
| 7 |

|
EtOH | 90 |
| 8 |

|
EtOH | 89 |
| 9 |

|
EtOH | 95 |
| 10 |

|
EtOH | 92 |
| 11 |

|
EtOH | 98 |
| 12 |

|
iPrOH | 90 |
| 13 |

|
iPrOH | 90 |
| 14 |

|
iPrOH | 99 |
| 15 |

|
iPrOH | 76 |
| 16 |

|
iPrOH | 81 |
| 17 |

|
t BuOH | 17b |
Changing the alcohol substrate from a primary (ethanol) to a secondary alcohol (2-propanol) did not have a significant effect on product yield, except slightly when using ortho-substituted aryl iodide substrates (Table 2, entries 12–16). However, moving to a tertiary alcohol (tert-butanol) was not successful, a low product yield being obtained presumably due to steric bulk (Table 2, entry 17).
We wanted to probe the scalability of the reaction. To do this, we screened the ethoxy- and isopropoxycarbonylation reaction with increasing quantities of iodobenzene. The results are shown in Table 3. With ethanol as a substrate, it is possible to scale the reaction to 4 mmol without a drop in product yield if the reaction time is extended from 20 min to 30 min (Table 3, entries 1–4). The drop in yield beyond this scale could be attributed in part to the fact that the CO concentration becomes too low. The parameters of the apparatus allow only a 10 atm initial gas load. However, given that the microwave unit can accommodate up to eight reaction vessels, it would be possible to prepare up to 32 mmol of a desired product in one run. With 2-propanol, moving to higher than the 1.5 mmol scale had a deleterious effect on product yield (Table 3, entries 5–8).
|
|
|||
|---|---|---|---|
| Entry | Alcohol | Scale/mmol | Product yield (%) |
| a Reactions were run in a sealed tube, pre-loaded with 10 bar CO, using the noted quantity of aryl halide, 15 mL of the desired alcohol, 0.1 mol% Pd(OAc), and 1.1 equiv. DBU. An initial microwave irradiation power of 1000 W was used, the temperature being ramped from r.t. to 125 °C and held until a total reaction time of 30 min had elapsed. | |||
| 1 | EtOH | 1.5 | 91 |
| 2 | EtOH | 3.0 | 93 |
| 3 | EtOH | 4.0 | 92 |
| 4 | EtOH | 5.0 | 78 |
| 1 | iPrOH | 1.5 | 92 |
| 2 | iPrOH | 3.0 | 83 |
| 3 | iPrOH | 4.0 | 75 |
| 4 | iPrOH | 5.0 | 64 |

In summary, using microwave heating it is possible to perform alkoxycarbonylation reactions of aryl iodides in reaction vessels pre-pressurized with carbon monoxide. Reactions can be performed using 0.1 mol% palladium acetate as catalyst with no additional ligand required. DBU proves to be the optimal base for the reaction. A range of aryl iodide substrates can be converted to the corresponding esters using this methodology. While ethanol and 2-propanol work well, tert-butanol proves less reactive. The potential for scale-up of the reaction has also been explored.
Experimental
General experimental
All aryl iodides were obtained from commercial suppliers and used without further purification. For the alcohol substrates: ethanol used was 200 proof; commercially available anhydrous 2-propanol was used; tert-butanol was distilled over potassium tert-butoxide. Reaction vessels were loaded with reagents without the need for exclusion of air. 1H- and 13C-NMR spectra were recorded at 293 K on a 400 MHz spectrometer. All product mixtures were worked-up using the specific procedure outlined below. Yields were determined by integration of NMR signals relative to an internal standard.Description and use of the microwave apparatus
A commercially available multimode microwave unit (Anton Paar Synthos 3000) was used for the reactions. The instrument is equipped with two magnetrons, with combined continuous microwave output power from 0 to 1400 W. Heavy-walled quartz reaction vessels (80 mL capacity, up to 60 mL working volume) were used. These vessels are dedicated for reactions at high pressure (up to 80 bar) and temperatures. The quartz vessels were capped with special seals and a protective PEEK cap and then were placed inside protecting air cooling jackets made of PEEK. The seals comprise a release valve that could be manually operated. The individual vessels were placed in an eight-position rotor and fixed in place by screwing down the upper rotor plate, and the rotor was finally closed with a protective hood. The vessels, once placed and secured in the rotor with the hood attached, were loaded with gas using a commercially available gas-loading kit, access being via a bayonet link. The vessels were evacuated and flushed with CO three times and then a pressure of 10 atm CO was adjusted at ambient temperature. The rotor was then placed into the microwave cavity. The temperature was monitored using an internal gas balloon thermometer placed in one reference vessel and additionally by exterior IR thermography. Pressure was monitored by a simultaneous hydraulic pressure-sensing device for all vessels, with recording of the highest pressure level and pressure increase. Reaction vessels were stirred by means of a rotating magnetic plate located below the floor of the microwave cavity and a Teflon-coated magnetic stir bar in the vessel. At the end of a reaction, any remaining pressure was vented by releasing the venting screw on each reaction vessel whilst still in the rotor, access being via frontal holes in the rotor lid.General experimental procedure
Acknowledgements
The American Chemical Society Petroleum Research Fund is thanked for funding (45433-AC1). Anton Paar GmbH is thanked for equipment support.References
- For book chapters see: (a) M. Mori, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, Wiley-Interscience, New York, 2002, vol. 2, pp. 2663 Search PubMed; (b) M. Beller, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 2nd edn, 2002, vol. 1, pp. 145–156 Search PubMed.
- For general reviews of palladium-mediated carbonylation chemistry see: (a) A. Zapf and M. Beller, Chem. Commun., 2005, 431 RSC; (b) R. Skoda-Földes and L. Kollár, Curr. Org. Chem., 2002, 6, 1097 CAS.
- For examples see: (a) W. Magerlein, A. F. Indolese and M. Beller, Angew. Chem., Int. Ed., 2001, 40, 2856 CrossRef CAS; (b) J. Albaneze-Walker, C. Bazaral, T. Leavey, P. G. Dormer and J. A. Murry, Org. Lett., 2004, 6, 2097 CrossRef CAS; (c) H. U. Blaser, M. Diggelmann, H. Meier, F. Naud, E. Scheppach, A. Schnyder and M. Studer, J. Org. Chem., 2003, 68, 3725 CrossRef CAS; (d) M. Z. Cai, H. Zhao and Y. X. Huang, J. Mol. Catal. A: Chem., 2005, 238, 41 CrossRef CAS; (e) W. Magerlein, M. Beller and A. F. Indolese, J. Mol. Catal. A: Chem., 2000, 156, 213–221 CrossRef CAS.
- For an example of the use of this chemistry on a multi-kg scale see: A. Zapf and M. Beller, Top. Catal., 2002, 19, 101 Search PubMed.
- (a) H. Neumann, A. Brennfuhrer, P. Grob, T. Riermeier, J. Almena and M. Beller, Adv. Synth. Catal., 2006, 348, 1255–1261 CrossRef CAS; (b) C. Jimenez-Rodriguez, G. R. Eastham and D. J. Cole-Hamilton, Dalton Trans., 2005, 1826 RSC.
- (a) A number of books on microwave-promoted synthesis have been published recently: Microwaves in Organic Synthesis, ed. A. Loupy, Wiley-VCH, Weinheim, 2006 Search PubMed; (b) C. O. Kappe and A. Stadler, Microwaves in Organic and Medicinal Chemistry, Wiley-VCH, Weinhiem, 2005 Search PubMed; (c) Microwave-Assisted Organic Synthesis, ed. P. Lidström and J. P. Tierney, Blackwell, Oxford, 2005 Search PubMed; (d) Microwaves in Organic Synthesis, ed. A. Loupy, Wiley-VCH, Weinheim, 2002 Search PubMed; (e) B. L. Hayes, Microwave Synthesis: Chemistry at the Speed of Light, CEM Publishing, Matthews NC, 2002 Search PubMed.
- For recent reviews see: (a) C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250 CrossRef CAS; (b) M. Larhed, C. Moberg and A. Hallberg, Acc. Chem. Res., 2002, 35, 717 CrossRef CAS; (c) A. Lew, P. O. Krutzik, M. E. Hart and A. R. Chamberlain, J. Comb. Chem., 2002, 4, 95 CrossRef CAS; (d) P. Lidström, J. P. Tierney, B. Wathey and J. Westman, Tetrahedron, 2001, 57, 9225 CrossRef CAS.
- O. Milijanic, K. P. C. Vollhardt and G. D. Whitener, Synlett, 2003, 29.
- E. Petricci, A. Mann, A. Schoenfelder, A. Rota and M. Taddei, Org. Lett., 2006, 8, 3725 CrossRef CAS.
- C. M. Kormos and N. E. Leadbeater, Synlett, 2006, 1663 CrossRef CAS.
- For an overview of the apparatus see: A. Stadler, B. H. Yousefi, D. Dallinger, P. Walla, E. Van der Eycken, N. Kaval and C. O. Kappe, Org. Process Res. Dev., 2003, 7, 707 Search PubMed.
- The use of this apparatus in the Diels–Alder reaction under an atmosphere of ethene has been reported: N. Kaval, W. Dehaen, C. O. Kappe and E. Van der Eycken, Org. Biomol. Chem., 2004, 2, 154 Search PubMed.
- For examples see: (a) O. Lagerlund and M. Larhed, J. Comb. Chem., 2006, 8, 4 CrossRef CAS; (b) X. Y. Wu and M. Larhed, Org. Lett., 2005, 7, 3327 CrossRef CAS; (c) X. Y. Wu, P. Nilsson and M. Larhed, J. Org. Chem., 2005, 70, 346 CrossRef CAS; (d) J. Wannberg and M. Larhed, J. Org. Chem., 2003, 68, 5750 CrossRef CAS.
- C. Ramesh, R. Nakamura, Y. Kubota, M. Miwa and Y. Sugi, Synthesis, 2004, 501.
- W. Magerlein, A. F. Indolese and M. Beller, J. Organomet. Chem., 2002, 641, 30 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photographs of the gas loading apparatus. See DOI: 10.1039/b614025d |
| This journal is © The Royal Society of Chemistry 2007 |