A highly effective one-pot synthesis of quinolines from o-nitroarylcarbaldehydes†
An-Hu
Li
*,
Eilaf
Ahmed
,
Xin
Chen
,
Matthew
Cox
,
Andrew P.
Crew
,
Han-Qing
Dong
,
Meizhong
Jin
,
Lifu
Ma
,
Bijoy
Panicker
,
Kam W.
Siu
,
Arno G.
Steinig
,
Kathryn M.
Stolz
,
Paula A. R.
Tavares
,
Brian
Volk
,
Qinghua
Weng
,
Doug
Werner
and
Mark J.
Mulvihill
*
Department of Cancer Chemistry, OSI Pharmaceuticals, Inc., 1 Bioscience Park Drive, Farmingdale, NY 11735, USA. E-mail: ali@osip.com; mmulvihill@osip.com; Fax: +1-631-845-5671; Tel: +1-631-962-0600
First published on 6th November 2006
Abstract
A highly effective one-pot Friedländer quinoline synthesis using inexpensive reagents has been developed. o-Nitroarylcarbaldehydes were reduced to o-aminoarylcarbaldehydes with iron in the presence of catalytic HCl (aq.) and subsequently condensed in situ with aldehydes or ketones to form mono- or di-substituted quinolines in high yields (66–100%).
The quinoline moiety is present as a substructure in a broad range of both natural and unnatural biologically active compounds, most notably within antimalarial agents.1 Due to such biological importance, quinoline derivatives have become the synthetic targets of many organic and medicinal chemistry groups,2 and new methods for constructing the quinoline ring appear regularly in the literature.3 Among these are new methodologies, modifications or improvements to the traditional Friedländer quinoline synthesis.4 The traditional Friedländer synthesis consists of two separate steps: the reduction of an o-nitroarylcarbaldehyde to an o-aminoarylcarbaldehyde and then condensation of the isolated o-aminoarylcarbaldehyde with a carbonyl compound under either basic or acidic conditions. A complicating factor for this stepwise procedure is the relative instability of the intermediate o-aminoarylcarbaldehyde, which can readily undergo self-condensation. This potential issue has prompted many laboratories to attempt to make improvements to this reaction,4a in particular through the use of one-pot reactions,3g,3k which negate the need for isolation of the intermediate aniline, are also less time- and labor-intensive than stepwise reactions, and usually afford higher yields.
A recent one-pot method, developed by Miller and McNaughton, uses SnCl2 as the reducing agent and ZnCl2 as the condensation promoter.3g Under these conditions a variety of substituted o-nitrobenzaldehydes were reacted with aliphatic ketones and reported to afford alkyl-derived quinoline products in high yields. As part of our ongoing drug discovery efforts, we required an efficient and practical method to access a variety of diverse 2-arylquinoline building blocks. We began by applying Miller's method to acetophenone but found it to be limited in application, affording only a trace amount of the desired aryl-derived quinoline product. Consequently, we embarked on establishing a new, one-pot procedure, which would be applicable for such aromatic substrates. Herein, we report a simple, efficient and practical one-pot quinoline synthesis using inexpensive and readily available reagents.
It is well-known that a nitro group can be reduced to an amino group using iron under acidic conditions. We found that o-nitrobenzaldehyde (1) was successfully and cleanly converted into o-aminobenzaldehyde (2) using 10 equivalents of iron in the presence of aq. HCl (20 mol%) in refluxing EtOH in just 30 min. The solids were removed by filtration and the filtrate was treated with ketone 3 and powdered KOH (3 eq.). After stirring at reflux for 40 min, 2-(4-pyridyl)quinoline (4) was obtained in quantitative yield (Scheme 1).
 | ||
| Scheme 1 | ||
This result encouraged us to explore the possibility of one-pot operations where intermediate 2 was not isolated. After several attempts, we developed a successful one-pot synthesis described hereafter. o-Nitrobenzaldehyde was reduced with 4.5 equivalents of iron powder in the presence of 5 mol% of HCl (aq.) in EtOH under reflux. The reduction was usually complete in 30–40 min (monitored by TLC), after which 1.0 equivalent of a ketone or aldehyde and 1.2 equivalents of KOH powder were added. After stirring under reflux for an additional 40–60 min, the condensation reaction was complete. A typical aqueous workup followed by chromatography over silica gel or recrystallization afforded the desired quinoline products in high yields.5 The procedure was successfully applied to a broad variety of ketones and aldehydes, the results from which are summarized in Table 1.

| Entry | Nitroaldehyde | Carbonyl compound | Product | Yield (%)a |
|---|---|---|---|---|
| a All reactions were carried out on a 1.0 mmol scale. The reaction times for the reduction and condensation stages were 40 min and 30 min, respectively, unless otherwise noted. All yields are isolated yields. See ref. 5 for a representative experimental procedure. b 60 min for reduction. c 2 h and 3 h for reduction and condensation, respectively. d 2 h for condensation. e Product was isolated as the HCl salt. f 5 and 48 h for reduction and condensation, respectively. g 8 eq. of iron and 16 mol% 0.1 N HCl (aq.) were used. 5 h and 15 h for reduction and condensation, respectively. h 1 h and 5 h for reduction and condensation, respectively. | ||||
| 1 | 1 |

|

|
99 |
| 2 | 1 |
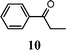
|

|
92b |
| 3 | 1 |

|

|
66c |
| 4 | 1 |

|

|
80 |
| 5 | 1 |

|
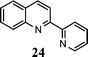
|
92 |
| 6 | 1 |

|
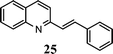
|
77 |
| 7 | 1 |

|
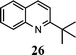
|
90d |
| 8 | 1 |
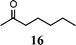
|

|
44 |
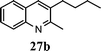
|
47 | |||
| 9 | 1 |

|
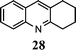
|
95 |
| 10 | 1 |
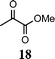
|

|
95e |
| 11 |
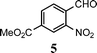
|
9 |
|
91e |
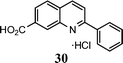
| 12 |

|
9 |
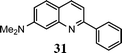
|
67f |
| 13 |

|
9 |
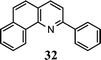
|
95g |
| 14 |

|
9 |

|
82 |
| 15 | 1 |

|

|
87h |
A broad range of o-nitroarylcarbaldehydes and carbonyl compounds was examined in our new one-pot procedure (Table 1). In comparison with Miller's acidic conditions,3g our procedure worked not only with aliphatic ketones but also with a wide variety of other ketones, including aromatic (entries 1–3, 11–14), heteroaromatic (entries 4 and 5), and α,β-unsaturated (entry 6) ones. As a result, 2-alkyl, 2-(het)aryl-, 2-phenylethenyl-, 2-carboxyl-, 2,3-dialkyl-, and 2-phenyl-3-methoxyquinolines can be prepared in good to excellent yields. It is especially noteworthy that even an aldehyde containing an active methylene can be successfully used for condensation (entry 15). This result further demonstrates the versatility of the reaction and allows for the preparation of 3-substituted quinolines, in contrast to the typical 2-substituted quinolines (entry 15 vs. entry 1). In this case, it appears that under these conditions the condensation stage is a rapid process and is able to effectively out-compete the self-condensation of phenylacetaldehyde.
The method described can also be used to prepare 2,3-disubstituted quinolines (entries 2, 3, 8 and 9). When an unsymmetrical ketone was used, a mixture of isomers was generated, which proved to be easily separable by silica gel column chromatography (entry 8). Use of a cyclic ketone afforded the respective tricyclic product (entry 9). Reaction of ketoester 18 afforded quinaldic acid in 95% yield (entry 10). 1-Nitronaphthalene-2-carboxaldehyde (7) resulted in a tricyclic heteroaromatic ring, such as benzo[h]quinoline (entry 13). From the heterocyclic o-nitroaldehyde 8, a substituted 1H-pyrazolo[4,3-b]pyridine was prepared (entry 14). o-Nitrobenzaldehydes with both electron-withdrawing and electron-donating substituents proceeded cleanly (entries 11 and 12) with the only exception being that a longer reaction time was necessary for the electron-rich dimethylamino case (entry 12). This one-pot procedure is also mild enough to allow a phenyl-substituted α,β-unsaturated ketone to be used as a reactant under basic conditions (KOH) in refluxing ethanol without significant competition from 1,4-Michael addition (entry 6).
In conclusion, we have developed a rapid, simple, and highly effective one-pot synthesis of both 2- or 3-mono-substituted and 2,3-disubstituted quinolines and also other bi- or tricyclic quinoline-derived heterocycles. The reaction proceeds with a wide variety of ketones and aldehydes, using inexpensive, readily available reagents and solvents that do not require oxygen- or moisture-free operations. The basic conditions are mild enough to tolerate substrates such as α,β-unsaturated Michael acceptors, as well as aldehydes and ketones prone to self-condensation. As such, this protocol adds to the Friedländer quinoline synthesis arsenal, providing another efficient means to synthesize many biologically active natural and unnatural quinoline-derived compounds.
Acknowledgements
We thank OSI Cancer Chemistry and especially Viorica Lazarescu and Raphael Rios for analytical support.References
- Recent reviews: (a) P. A. Stocks, K. J. Raynes and S. A. Ward, Antimalarial Chemotherapy, 2001, 235 Search PubMed; (b) N. C. Waters, G. S. Dow and M. P. Kozar, Expert Opin. Ther. Pat., 2004, 14, 1125 Search PubMed; (c) C. D. Fitch, Life Sci., 2004, 74, 1957 CrossRef CAS; (d) P. L. Olliaro and W. R. J. Taylor, J. Exp. Biol., 2003, 206, 3753 Search PubMed; (e) J. P. Michael, Nat. Prod. Rep., 2004, 21, 650 RSC.
- Some recent papers: (a) A. H. Abadi, G. H. Hegazy and A. A. El-Zaher, Bioorg. Med. Chem., 2005, 13, 5759 CrossRef CAS; (b) J. E. Charris, J. N. Dominguez, N. Gamboa, J. R. Rodrigues and J. E. Angel, Eur. J. Med. Chem., 2005, 40, 875 CrossRef CAS; (c) A. G. Tempone, A. C. M. P. Da Silva, C. A. Brandt, F. S. Martinez, S. E. T. Borborema, M. A. B. Da Silveira and H. F. De Andrade, Jr, Antimicrob. Agents Chemother., 2005, 49, 1076 CrossRef CAS.
- (a) J. E. Na, K. Y. Lee, D. Y. Park and J. N. Kim, Bull. Korean Chem. Soc., 2005, 26, 323 CAS; (b) C. S. Cho, H. J. Seok and S. C. Shim, J. Heterocycl. Chem., 2005, 42, 1219 CrossRef CAS; (c) J. Wang, X. Fan, X. Zhang and L. Han, Can. J. Chem., 2004, 82, 1192 CrossRef CAS; (d) E. Ronne, K. Olsson and S. Grivas, Synth. Commun., 1994, 24, 1363 CrossRef CAS; (e) K. Motokura, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2004, 45, 6029 CrossRef CAS; (f) V. Bailliez, L. El Kaim and V. Michaut, Synth. Commun., 2004, 34, 109 CrossRef CAS; (g) B. R. McNaughton and B. L. Miller, Org. Lett., 2003, 5, 4257 CrossRef CAS; (h) B. Jiang and Y.-G. Si, J. Org. Chem., 2002, 67, 9449 CrossRef CAS; (i) K. Mogilaiah and C. S. Reddy, Synth. Commun., 2003, 33, 3135 CrossRef; (j) Y. Hsiao, N. R. Rivera, N. Yasuda, D. L. Hughes and P. J. Reider, Org. Lett., 2001, 3, 1101 CrossRef CAS; (k) H. Eckert, Angew. Chem., 1981, 93, 216 CrossRef CAS.
- (a) A review: C.-C. Cheng and S.-J. Yan, Org. React., 1982, 28, 37 Search PubMed; (b) A recent paper on mechanism: J. M. Muchowski and M. L. Maddox, Can. J. Chem., 2004, 82, 461 Search PubMed.
- Representative experimental procedure (entry 9, Table 1): To a solution of o-nitrobenzaldehyde (1, 151 mg, 1.0 mmol) in ethanol (3 mL) was added iron powder (<10 µm, Aldrich, 223 mg, 4.0 mmol) followed by 0.1 N aq. HCl (0.5 mL, 0.05 mmol), and the resulting mixture was vigorously stirred at 95 °C (oil bath) for 40 min. TLC analysis revealed that the reduction reaction was complete, so cyclohexanone (17, 0.10 mL, 1.0 mmol) and powdered KOH (67.3 mg, 1.2 mmol) were then added successively in portions (Caution! Potential exotherm; add KOH slowly). The reaction mixture was stirred at 95 °C for 30 min, then cooled to rt, diluted with CH2Cl2 (50 mL), and filtered through a Celite pad. The filtrate was washed with water (10 mL) and the aqueous phase was back-extracted with CH2Cl2 (2 × 15 mL). The combined organic phases were dried over MgSO4, filtered, and concentrated in vacuo. The crude material was purified by chromatography over silica gel (EtOAc–hexane 1 : 4) to afford 173 mg of 1,2,3,4-tetrahydroacridine (28, 95%).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/b613775j |
| This journal is © The Royal Society of Chemistry 2007 |