Multistate reaction kinetics of 6-hydroxy-4′-(dimethylamino)flavylium driven by pH. A stopped-flow study
Received 20th July 2006, Accepted 6th October 2006
First published on 16th November 2006
Abstract
The synthetic flavylium salt 6-hydroxy-4′-(dimethylamino)flavylium hexafluorophosphate displays a set of pH-driven chemical reactions in aqueous solutions, involving the formation of hemiketal species and chalcones with cis and trans configurations. Such reactions were studied by steady-state and transient UV-Vis spectroscopy and by stopped-flow techniques. A novel and more generalized kinetic scheme is developed, in order to take account of possible acid/base pairs that occur in the network of chemical reactions as the pH is changed. It is found that the formation of the hemiketal species by hydration of the flavylium is slow, and it is not possible to isolate each process that leads to the formation of the cis-chalcone (hydration and tautomerization reactions). The cis/trans isomerization reaction of cis-chalcone is slow, and the system takes several hours to reach equilibrium after a pH jump at room temperature. In basic conditions, negatively charged trans-chalcones are dominant. Comparison with other flavylium compounds shows that the hydration process is affected mainly by the amino group, while the hydroxyl group influences the tautomerization and isomerization reactions.
Introduction
The multistate pH-driven reactions of flavylium dyes produce systems in which complex logical functions may be carried out in solution.1 This is possible because of the wide range of two-state processes that appear by applying stimuli, such as a pH jump or a photochromic process, to the system. Flavylium compounds, as well as other molecules like natural anthocyanins, present a 2-phenylbenzopyrylium core which develops into several species, such as the hemiketal B obtained by hydration in the 2-position of the flavylium cation, a cis-2-hydroxychalcone Cc formed by tautomerization of B, and a trans-2-hydroxychalcone Ct obtained by isomerization of Cc (Fig. 1).2,3 Suitable substituents might be introduced into the general skeleton of the flavylium compounds in several positions, influencing greatly the reactivity of the compounds.1–16 The substitution by hydroxyl or methoxy in position 4′ and the absence of electron donor groups in position 7 are particularly important, because it has been shown that it leads to a cis/trans barrier, allowing these compounds to undergo write–lock–read–unlock–erase photochromic cycles, and perform as XOR and XNOR logic gates (multistate/multifunctional systems).1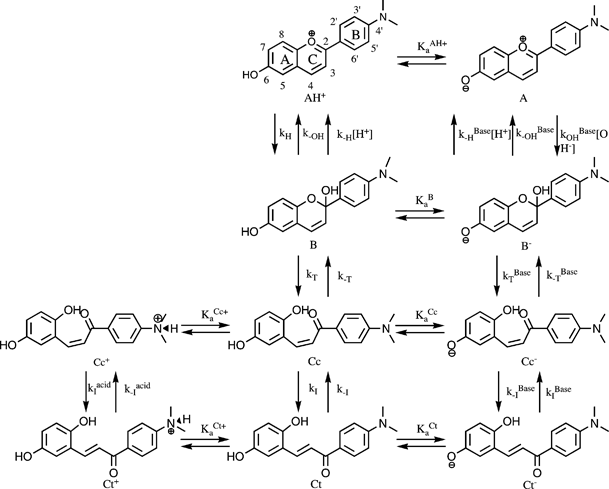 |
| | Fig. 1 pH-Driven chemical reactions of 6-hydroxy-4′-(dimethylamino)flavylium. | |
This work deals with 6-hydroxy-4′-(dimethylamino)flavylium hexafluorophosphate, a compound with a dimethylamino group at the 4′-position and a hydroxyl group at the 6-position (see Fig. 1). This compound can be made to undergo a wide range of chemical reactions just by changing the pH, and it is important to understand a priori the effects of the substituents on this network.1 The presence of the dimethylamino group has already been studied,6,12–14,16 and it was found that the formation of B, through the hydration of AH+, is slow due to an extended conjugation between the benzopyrylium and the dimethylaniline rings, giving rise to a planar flavylium ion. The presence of the hydroxyl group at the 6-position opens up new possibilities, increasing greatly the number of states that might be achieved by pH jumps (see Fig. 1) because of all the hydration and tautomerization reactions that might occur on the conjugated base.17 The existence of a new pathway for the conversion of flavylium compounds into chalcones will affect the kinetics greatly; however it has been found that the hydration of A is negligible. The reaction kinetics and the thermodynamics are affected by this increase, and several assumptions and simplifications that are usually made to describe these results cannot be applied here, which increases the complexity of the problem. Instead, a more general scheme is developed, which can be used in many flavylium systems, considering that acid/base reactions reach equilibrium immediately. The multistate reaction then transforms into a 3-state reaction scheme, which is solved by the matrix method.18,19 With the kinetic analysis and the equilibrium results, it was possible to obtain all the kinetic and equilibrium constants, and these are compared with the data available for other flavylium compounds.
Thermodynamics and kinetics
As mentioned in the introduction, flavylium compounds display a wide range of chemical reactions driven by pH, caused not only by acid/base equilibrium, but also because the hydration of the flavylium ring leads to the formation of a hemiketal compound (B) and H+. B ultimately will open and transforms into the cis-chalcone (Cc) by tautomerization and later into the trans-isomer (Ct) by cis/trans isomerization.1–3 The basic partners of AH+, B, Cc and Ct are A, B−, Cc− and Ct−, which are formed due to deprotonation of the hydroxyl group (see Fig. 1). Many flavylium compounds do not have such a group, and therefore will not undergo any of the reactions on the right-hand side of Fig. 1, which simplifies the thermodynamic and kinetic analysis. On the other hand, most of the reactions occur on totally different timescales. The formation of B and Cc from AH+ takes place normally within seconds, or fractions of a second, while the cis/trans isomerization generally occurs on a very long timescale (several hours or more). All the acid/base equilibria are virtually instantaneous when compared with the other reactions presented here.With the assumption that the acid/base reactions are instantaneous, the molar fractions of each species will be given by:
| |  | (1-A) |
| |  | (1-B) |
| |  | (1-C) |
| |  | (1-D) |
| |  | (1-E) |
| |  | (1-F) |
Eqn (1) neglects acid/base reactions that produce Cc
2− and Cc
+ species (it is assumed that the kinetics is studied at moderate pH). The same equations may also be written for the Ct acid/base equilibria. Since the isomerization reaction takes place on a long timescale, only the reactions from AH
+ to Cc will be considered in the following kinetics. The following pseudo-rate constants may be defined.
| |  | (2-A) |
| | 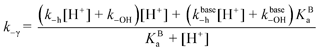 | (2-B) |
| |  | (2-C) |
| |  | (2-D) |
The following set of differential equations describes the kinetics of the reaction of AH
+/A to give Cc/Cc
−:
| | 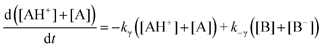 | (3-A) |
| |  | (3-B) |
| |  | (3-C) |
The solution of
eqn (3) is given by a sum of exponentials, where the solutions of the time constants are given by the following determinant:
18,19| |  | (4) |
and therefore
| |  | (5) |
The reciprocals of
λ give the relaxation times of the kinetic traces obtained by pH jumps of stopped-flow experiments.
2,3,10 Because
λ1 = 0, it turns out that the decays are biexponential even though the determinant is 3 × 3. If no acid/base equilibria are observed for AH
+, B and Cc,
eqn (5) reduces to:
| |  | (6) |
In certain conditions (acidic media, basic media or low tautomerization rate constants)
eqn (5) gives:
| |  | (7) |
Unfortunately,
eqn (7) cannot be applied to the whole pH range in some cases, and if one desires to describe the whole set of data,
eqn (5) is necessary. Yet, in many cases the approximation of
eqn (7) is enough to describe experimental data in acidic conditions, because a linear relationship with [H
+] is observed, which therefore can be used to obtain the rate constants. As will be shown below, however, the pH dependence is sometimes difficult to linearize for certain flavylium compounds, and a non-linear data fit with
eqn (5) is required in order to obtain the rate constants. After such a procedure it may be proven that
eqn (7) is enough to describe the data, but such knowledge is only known
a posteriori. If the use of
eqn (7) is valid, some approximations can be made and some useful equations found. If [H
+] ≫
KBa (a situation that might be common over a wide pH range, because p
KBa is expected to be above 9):
| | 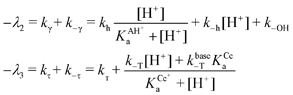 | (8) |
Eqn (8) shows how certain rate constants may be directly obtained from experiments. If
k−OH is not negligible, a multilinear regression of
λ2vs. [H
+] and [H
+]/(
KAH+a + [H
+]) (if
KAH+a is known) would give rise to a non-zero value at the intercept with [H
+] = 0. If one is sure that
eqn (7) is applicable, than
eqn (8) is a practical way to get the results needed at pH < 10.
Also, some processes within the scheme have been proven to be quite slow or almost non-existent, as in the case of A hydration, represented by kbase−h![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) [H+]. If this term is equal to zero, a simplification occurs, and the same reasoning applies to all other processes on this network of chemical reactions. It is therefore important to understand whether or not there are any processes that can be neglected. The quality of the experimental data is also rather critical, in order to correctly catch the shape of the time decay constants vs. pH curves.
[H+]. If this term is equal to zero, a simplification occurs, and the same reasoning applies to all other processes on this network of chemical reactions. It is therefore important to understand whether or not there are any processes that can be neglected. The quality of the experimental data is also rather critical, in order to correctly catch the shape of the time decay constants vs. pH curves.
Once the thermodynamic equilibrium is reached, with the establishment of the cis/trans isomerization, normally only AH+, A, Ct and Ct− species remain, depending on the pH. As a first approximation, [AH+] may be obtained from the following equation:4,5
| |  | (9) |
in which
CAH is the total concentration of flavylium species,
Ka′ is an apparent acidity constant given by:
| | | Ka′ = KAH+a + KH + KHKT + KHKTKI | (10) |
and the equilibrium constants (easily obtained with a pH titration) are:
| | 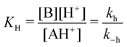 | (11-A) |
| |  | (11-B) |
| |  | (11-C) |
However, within the scheme shown in Fig. 1, this approach is no longer valid if one desires to explain the whole set of data. KH, as defined in eqn (11-A), is not a constant because it will depend on [H+]. The acid/base equilibria for B, Cc and Ct should be considered as well. [AH+] should be obtained by the following equation:
| |  | (12) |
where:
| | | Ka″ = KBaKH + KCcaKHKT + KCtaKHKTKI | (13) |
and
| |  | (14) |
If the appearance of Cc
2− and Ct
2− are also considered,
eqn (12) must change as well, in order to accommodate these species into the description. The change might be important at high pH when these species appear and hence shift the equilibrium towards the formation of the chalcone. The full description is then given by:
| |  | (15) |
where:
| | 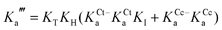 | (16) |
The same reasoning is also valid if Ct
+ and Cc
+ (and AH
2+ and BH
+, which were not considered because such species appear at very low pH values, below 1) are considered in the scheme, but such species are unlikely to appear because under acidic conditions only the flavylium salt remains. Nevertheless, one has to realize that experimentally it is [AH
+]
vs. pH that is obtained by probing a wavelength at which AH
+ (and almost nothing else) absorbs light, and under such conditions
eqn (9) is sufficient. Only if species like Ct
− or Ct
2− are being probed will
eqn (12) or
eqn (16) be more appropriate.
Results and data analysis
Structure
6-Hydroxy-4′-(dimethylamino)flavylium has a structure similar to 4′-aminoflavylium salt, which has been previously studied by X-ray diffraction.12 X-Ray analysis of this salt showed an almost planar molecule with a resonance form in which the amino group participates in the π molecular orbital (see Scheme 1). Therefore the positive charge is distributed and the hydration reaction in which the hemiketal species is formed is less favorable. AM1 semi-empirical calculations for the present case-study suggest that these conclusions may be extended to 6-hydroxy-4′-(dimethylamino)flavylium (see Fig. 2). Table 1 compares three different flavylium compounds and shows how crucial the dimethylamino group bonded to C4′ is in order to put the phenyl ring planar with the rest of the molecule. An important double bond character is also found in C4′–N and C2–C1′ bonds for the dimethylamino derivatives, which is absent for 6-hydroxyflavylium and the hemiketal species. The double bound character does not substantially change the C2 charge, but nucleophilic attack on this carbon will be more difficult. In addition, the amino group is not easily protonated, and will have a low pKa. |
| | Scheme 1 Resonance structure of 6-hydroxy-4′-(dimethylamino)flavylium | |
 |
| | Fig. 2 Structure of 6-hydroxy-4′-(dimethylamino)flavylium obtained by AM1 semi-empirical calculations. | |
Table 1 AM1 semi-empirical calculations
| | C2 charge | O1–C2–C1′–C2′ bond torsion | C3′–C4′–N–C bond torsion | C2–C1′ bond order | C4′–N bond order | C2–O1 bond order |
|---|
| 6-Hydroxy-4′-(dimethylamino)flavylium |
| AH+ | 0.28 | 0.6° | 2.5° | 1.24 | 1.28 | 1.17 |
| A | 0.11 | 0.8° | 3.7° | 1.04 | 1.12 | 1.21 |
| B | 0.29 | 26.1° | 27.9° | 0.93 | 1.06 | 0.91 |
| B− | 0.30 | 28.1° | 11.4° | 0.91 | 0.97 | 0.97 |
| |
| 4′-Dimethylaminoflavylium |
| AH+ | 0.28 | 0.6° | 2.5° | 1.24 | 1.28 | 1.15 |
| B | 0.29 | 26.1° | 28.0° | 0.93 | 1.06 | 0.91 |
| |
| 6-Hydroxyflavylium |
| AH+ | 0.28 | 20.5° | — | 1.08 | — | 1.22 |
| A | 0.06 | 22.0° | — | 1.02 | — | 1.21 |
| B | 0.28 | 26.4° | — | 0.93 | — | 0.91 |
| B− | 0.30 | 28.3° | — | 0.92 | — | 0.97 |
| |
| 4′-Aminoflavylium (X-ray data)12 |
| AH+ | — | 2.2° | — | — | — | — |
Electronic absorption spectra – equilibrium
The electronic absorption spectra exhibit a strong absorbance at λ = 540 nm in acidic aqueous solutions (see Fig. 3). Such a band is characteristic of other flavylium compounds and is assigned to AH+.11 As the pH increases, this band gradually disappears, reflecting the disappearance of AH+. On the other hand, a new transition appears at 410 nm, which is assigned to the formation of chalcone species (both Cc and Ct). For pH > 10 almost all absorbance at 540 nm disappears, while the shape of the absorption spectra between 300 and 500 nm shows the acid/base equilibria of the chalcone species. A plot of the absorbance at 540 nm as a function of pH can be fitted with eqn (8), giving rise to a pKa′ of 6.4. This already gives an upper limit for the value of KAH+a. Also a separated absorption band from A is not observed, and the chalcone species have overlapping spectra. This does not enable a clear-cut description of the system to be formulated just by examining the electronic absorption spectra, and other experiments are required. |
| | Fig. 3 Absorption spectra of aqueous solutions of the 6-hydroxy-4′-(dimethylamino)flavylium compound (10 µM) as a function of pH (pH = 2.3 to 10.9). The spectra were run after the solutions were left to reach equilibrium in the dark for 48 h. Insert shows absorption spectra in the pH range 7.9–10.9. | |
Electronic absorption spectra – AH+/A equilibrium
Since the hydration of AH+ occurs on a rather long timescale, a pH jump from an acidic solution containing AH+ to a pH until about 8 will instantaneously produce an acid/base equilibrium. By recording the absorption spectra after this jump (around 30 s after) it is possible to approximately capture this equilibrium and therefore to obtain KAH+a and the absorption spectrum of A. These results are shown in Fig. 4, and it is concluded that KAH+a = 1.9 × 10−8 M. The isosbestic point at 560 nm is ill-defined because at higher pH there is already some hydration/tautomerization occurring. |
| | Fig. 4 Absorption spectra of aqueous solutions of the (5 µM) 6-hydroxy-4′-(dimethylamino)flavylium compound as a function of pH between 1.7 and 9 taken 30 s after a pH jump from 1. Insert shows absorption at λ = 540 nm. | |
Transient absorption spectra – UV-Visible spectrophotometry
The absorption spectra after a pH jump from 1 to 8.2 were measured as a function of time (see Fig. 5). Conversion of AH+ to A is observed during the mixing time of the sample: even after 2 minutes the absorption spectra of A peaked at 560 nm. Afterwards, the absorption at 560 nm decreases gradually, while a new band appears around 400 nm. A clear isosbestic point is not observed, so again it is not possible to distinguish different species within the spectra. However, it is possible to fit the data with a biexponential decay, in which one of the components is due to the cis/trans isomerization with a relaxation time of 3.6 hours (see Table 2). The other component is in fact a mixture of two components containing both hydration and tautomerization processes, which could be resolved by stopped-flow experiments, which have a much better time-resolution. |
| | Fig. 5 Transient absorption spectra between t = 2 min and 20 h after a pH jump from 1 to 8.2. After 3 h, all the flavylium species (AH+ and A) have disappeared, while Ct appears on a long timescale (more than 5 h). | |
Table 2 Isomerization rate constant at 293 K
| pH | kI/10−5 s−1 |
|---|
| 7.7 | 9.4 |
| 8.2 | 6.1 |
| 8.4 | 7.8 |
Stopped-flow – direct pH jumps
Starting from acidic solutions (pH ≈ 1), only AH+ is present. By a sudden increase of pH, a mixture of AH+/A is immediately achieved on the timescale of a stopped-flow experiment: even after 3 ms the spectrum is shifted to the wavelength characteristic of A (see Fig. 6). Therefore two processes are observed: the hydration of AH+, which generates the hemiketal compound B, and the tautomerization of B with the consequent ring opening, leading to the formation of the chalcone Cc. Nevertheless, such a picture is simplistic, because the reverse reactions back to AH+ also occur. However, as shown in eqn (5), biexponential kinetics are expected even when all these processes are taken into account, and, as shown in Fig. 6, this is what is observed experimentally.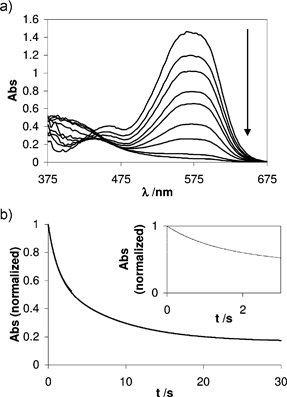 |
| | Fig. 6 Direct pH jump from 1 to 10 of a solution measured by stopped-flow. a) Time-resolved spectra for time delays of 3 ms, 0.5, 1, 2, 3, 6, 10, 20 and 30 s; b) Bleaching at λ = 545 nm. | |
Stopped-flow – reverse pH jumps
Starting off from pH = 7, four hours after the sample preparation, a mixture of AH+, A, Cc, Ct and B is expected at the beginning. The pH jump to acidic conditions leads to the formation of AH+, because of the reverse chemical reactions that take place. At the end, only AH+ and Ct (or Ct+) species will remain. Fig. 7 shows that A is immediately converted to AH+ after a pH jump from 7 to 1.6. Afterwards the absorption at λ = 540 nm increases with biexponential kinetics, and some minutes later only AH+ and Ct (or Ct+) species remain. The amplitude of the signals from reverse pH jumps is considerably lower than from direct pH jumps. The consequences are kinetic traces with much more noise, making them more difficult to analyze. As seen in Fig. 8, the errors associated with the decay constants are higher. |
| | Fig. 7 Reverse pH jump from 7 to 1.6 of a solution measured by stopped-flow. a) Time-resolved spectra for time delays of 8 ms, 80 ms, 0.5, 1, 12 and 60 s; b) Absorption increase at λ = 545 nm; the white line is a biexponential fit. | |
![a) pH dependence of the constants obtained from direct and reverse pH jumps with the stopped-flow technique, and the fit with eqn (5); b) Curves 1 and 2 from graph a), and kγ + k−γ[H+] (curve 3) and kτ + k−τ (curve 4) (see eqn (7)).](/image/article/2007/OB/b610466e/b610466e-f8.gif) |
| | Fig. 8 a) pH dependence of the constants obtained from direct and reverse pH jumps with the stopped-flow technique, and the fit with eqn (5); b) Curves 1 and 2 from graph a), and kγ + k−γ[H+] (curve 3) and kτ + k−τ (curve 4) (see eqn (7)). | |
Rate constants and equilibrium – full description
Fig. 8 shows a fit of the pH dependence of the constants obtained from stopped-flow experiments using eqn (5). This dependence shows an unusual dependence, especially for pH values between 5 and 10, where the kinetics is rather slow for the second component. The fitting results in the following values:
kbaseh, kbase−h, kOH, kbase−OH and kbase−t converge to meaningless values, suggesting that A is much less reactive than AH+. Also, the approximation of eqn (7) is only valid for pH values above 11 or lower than 1 (see Fig. 8b). Therefore, this is a system where each component of the kinetics may be addressed to one specific reaction (hydration or tautomerization) only under rather extreme pH conditions.It is now possible to calculate KI from Ka′ and KCta from Ka″ from the absorption spectra at equilibrium conditions. It is even possible to attain an estimation of KCt−a using eqn (14), since at certain wavelengths (between 420 and 290 nm) the absorption spectra show variations at pH values above 8 (see Fig. 3 and Fig. 9). This is an indication of the presence of Ct2− and/or Cc2− species at pH values above 8, and therefore it is possible to use eqn (15). However, it is not possible to distinguish between the two species, unless it is assumed that [Cc2−] is negligible, in which case only an indication of KCt−a is obtained.
 |
| | Fig. 9 Absorption variation under equilibrium conditions at different wavelengths fitted with eqn (18). | |
To fit the experimental data at various wavelengths, a general expression is required. According to the Lambert–Beer law, the absorption at a given wavelength is the sum of the absorptions of each of the species in solution. In this system there are many molecules in solution (AH+, A, B, B−, Cc, Cc−, Cc2−, Ct, Ct− and Ct2−), and each molecule will have an extinction coefficient ε for every wavelength. Discarding certain molecules that will exist in small amounts, like B−, eqn (17) should describe the experimental results.
| |  | (17) |
However, not all variables can be obtained, so it is better to work with this simplified expression described by
eqn (18), where
ε′,
ε″ and
ε‴ are related to the extinction coefficients.
| |  | (18) |
The fitting results (see Fig. 9) are given by:
| KCt−aKCtaKI + KCc−aKCca = 6.64 × 10−14 M2 |
If one considers the amount of Cc
2− to be negligible, one obtains
KCt−a = 2.0 × 10
−9 M (p
KCt−a = 8.7).
Discussion
Formation of the hemiketal
The results obtained show a rather slow hydration of AH+ species (kh = 0.005 s−1), which is almost the limiting step of the set of chemical reactions described in Fig. 1. Once B is formed, it readily reacts to form AH+ or Cc, at almost equal rates (kT = 0.30 s−1 and k−OH = 0.48 s−1), and under acidic conditions only AH+ is formed (k−h = 33.1 M−1 s−1). KH, which should be considered as an equilibrium parameter instead of a constant (see eqn (14)), decreases strongly with pH, acquiring very low values such as 10−10 M.The instability of B is explained by the double bond character of C2–C1′ (see Table 1), which hinders the nucleophilic attack by H2O. Such behavior has been observed in other flavylium compounds bearing an amine group on the 4′-position,12 and in 7-hydroxy-4′-(dimethylamino)flavylium.13 Therefore, electron-donating groups at such positions reduce the formation of B, slowing down all the kinetics of the scheme in Fig. 1. On the other hand, A is much less reactive than AH+, a result that has also been observed previously. Only by nucleophilic attack of A with OH− is it possible to form the hemiketal species, and therefore in basic conditions the reaction is catalyzed by OH−.
Formation of the chalcones
Kinetically, the formation of cis-chalcones (Cc) occurs after the formation of hemiketal species, and the cis/trans isomerization takes some hours to proceed, and therefore Ct species are not observed within the timescale of stopped-flow experiments. But, once equilibrium is reached, there is a rich chalcone pH-dependent chemistry. The mole fraction distribution of the several species can be calculated following a procedure reported in the literature1,11 (see Fig. 10), and it is found that even at pH 3 a significant amount of Ct exists in solution. Cc− appears in large quantities around pH 8, while for pH > 9 Ct2− (and perhaps Cc2−) becomes dominant. It is not possible to be certain of the presence of either Cc2− or Ct2− at such high pH with the present set of data, but, unless both species have a similar pKa, it is likely that only Ct2− is dominant. Cc2− should be much less stable than Cc− due to the hydrogen bond that should be formed in Cc−.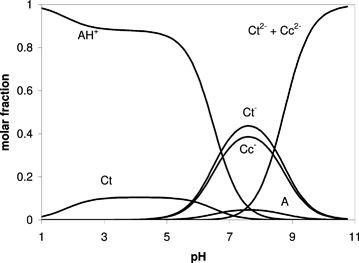 |
| | Fig. 10 Molar fraction distribution at the equilibrium pH for 6-hydroxy-4′-(dimethylamino)flavylium species. | |
Comparison with other flavylium compounds
Table 3 compares the kinetic and equilibrium constants of different and related flavylium compounds. All reactions depend strongly on the presence of the substituents. The amino group considerably decreases the rates of hydration and dehydration, presumably due to the planar structure and extended π conjugation of the AH+ species. The hydroxyl group, which is an electron-donor as well, also reduces the hydration rate, but in a less pronounced way. The presence of an electron-acceptor group like NO2, on the other hand, has an opposite effect. For 4′-hydroxy-6-nitroflavylium, a kh of 9 s−1 was obtained,5g a value that is 3 orders of magnitude higher than for 4′-hydroxyflavylium (kh = 8.9 × 10−2 s−1),5c another flavylium compound with a planar structure.
Table 3 Equilibrium and kinetic constants for different synthetic flavylium compounds
| Substituent | Noneb | 6-OHc | 4′-Dimethylaminod | 7-Diethylamino-4′-OHe | 7-OH-4′-Dimethylaminof | 6-OH-4′-Dimethylamino |
|---|
| Calculated using eqn (10). Ref. 3 and 11. Ref. 17. Ref. 12. Ref. 14. Ref. 13. |
|---|
| KH/M | 9.8 × 10−4 | 1 × 10−4 | 2.8 × 10−9 | — | 2.5 × 10−6 | 2.9 × 10−8a |
| KT | 0.06 | 0.57 | 21 | — | — | 0.47 |
| KI | 400 | 25 | 130 | — | >4000 | 25 |
| kh/s−1 | 4.6 | 0.09 | 2.8 × 10−5 | 1.5 × 10−4 | 0.02 | 0.005 |
| k−h/M−1 s−1 | 4.7 × 103 | 900 | 1000 | — | 8000 | 33.1 |
| kt/s−1 | 0.11 | — | 80 | — | — | 0.30 |
| k−t/s−1 | 1.78 | — | 3.7 | — | — | 0.64 |
| ki/s−1 | 4.1 × 10−4 | 1 × 10−4 | 4.8 × 10−6 | — | 0.4 | 7.8 × 10−5 |
| k−i/s−1 | 1.1 × 10−6 | — | 3.7 × 10−8 | 1 × 10−2 | <1 × 10−4 | — |
| kbaseOH/M−1 s−1 | — | — | 9.3 × 104 | 3.5 | 1.4 | 610 |
| k−OH/s−1 | 4.9 × 10−6 | — | — | — | — | 0.48 |
| kbaset/s−1 | — | — | — | — | — | 0.17 |
Such an effect is much less pronounced on the tautomerization and isomerization reactions: remarkably, the isomerization is almost identical on the 6-hydroxy- and 6-hydroxy-4′-(dimethylamino)- compounds, and the same seems to happen for the tautomerization reaction. Thus it seems that for these reactions the hydroxyl group at C6 has a stronger influence, perhaps because these reactions proceed from a molecule where the two rings are electronically decoupled. Therefore, rather than being an intermediate case, the chemistry of 6-hydroxy-4′-(dimethylamino)flavylium resembles 4′-aminoflavylium for the hydration reaction, while for the tautomerization and isomerization it resembles 6-hydroxyflavylium.
These two aspects provide, therefore, a framework in which the design of new flavylium compounds may be directed in order to obtain desirable chromic properties. Knowledge of how substituents affect the reaction kinetics is as crucial as the understanding of solvent effects. Qualitative predictions may then be complemented by semi-empirical calculations, in order to obtain the ideal pH-dependent rate constants for the network of chemical reactions of these compounds.
Final comments
The flavylium salt 6-hydroxy-4′-(dimethylamino)flavylium hexafluorophosphate, which has a wide array of pH-driven chemical reactions in aqueous solutions, was synthesized. In order to understand the chemistry of this type of compound, a generalized kinetic scheme is required. This leads to a better understanding of the relationship between chemical structure and reactivity, because it is possible to access many kinetic rate constants. In the present case, a comparison with other flavylium compounds shows that the hydration process is affected mainly by the amino group, while the hydroxyl group influences the tautomerization and isomerization reactions. This deduction is helpful in order to design better flavylium compounds for applications like information storage. Ultimately, the work developed here might also be applied in photochromic or electrochromic devices, enabling a framework in which compounds with a more complicated chemical structure might also be used in a quantitative way.Experimental
Synthesis
All reagents and solvents used were of analytical grade. NMR spectra were run on a Bruker AMX 400 instrument, MS spectra were run on a Micromass GCT machine, and elemental analyses were obtained on a Thermofinnigan Flash EA 1112 series device.6-Hydroxy-4′-(dimethylamino)flavylium hexafluorophosphate
This was prepared by condensation of 4′-(dimethylamino)acetophenone (0.69 g, 5 mmol) and 2,5-dihydroxybenzaldehyde (0.82 g, 5 mmol), on the basis of a method initially described by Robinson and Pratt,20 and later developed by Michaelidis and Wizinger,21 using concentrated sulfuric acid instead of gaseous hydrogen chloride. The reagents were dissolved in acetic acid (10 ml), and 98% (w/w) sulfuric acid (2.5 ml) was then added, keeping the temperature below 60 °C. The solution became red and was stirred overnight. Addition of ethyl acetate led to precipitation of a brown solid, which was filtered and washed with ethyl ether. A yield of 51% was obtained. The solid may be dissolved in acetic acid and reprecipitated as its hexafluorophosphate salt by adding 65% (w/w) HPF6. Elemental analysis: exp. (calc. for C17H16NO2PF6·0.5H2O; FW = 420.29 g mol−1) C 48.39% (48.58%), N 3.44% (3.33%), H 4.44% (4.08%). MS-FD: m/z 266.1 [M − PF6]+ (100%). 1H NMR (400 MHz, CD3CN/DCl, 300 K, AH+ species): δ/ppm 3.23 (s, 6H, N(CH3)2), 6.92 (d, 2H, 3J = 8.8 Hz, H3′ + H5′), 7.22 (d, 1H, 4J = 2.8 Hz, H5), 7.43 (dd, 1H, 3J = 9.1 Hz, 4J = 2.8 Hz, H7), 7.77 (d, 1H, 3J = 9.1 Hz, H8), 7.97 (d, 1H, 3J = 9.4 Hz, H3), 8.17 (d, 2H, 3J = 8.8 Hz, H2′ + H6′), 8.37 (d, 1H, 3J = 9.4 Hz, H4). 13C NMR (100 MHz, CD3CN/DCl, 300 K, AH+ species): δ/ppm 41.2 (N(CH3)2), 112.9 (C5), 114.7 (C3′ + C5′), 115.5, 117.6 (C3), 120.3 (C8), 124.6, 126.3 (C7), 134.2 (C3′ + C5′), 147.6 (C4), 149.8, 157.5, 158.2, 171.0.Measurements
All experiments were carried out in aqueous solution. The pH was adjusted by addition of HCl and NaOH, or buffer, and was measured in a Meterlab pHM240 pH meter from Radiometer Copenhagen. UV-Vis absorption spectra were recorded in a Shimadzu UV2501-PC spectrophotometer. The stopped-flow experiments were performed in a SFM-300 spectrophotometer, controlled by a MPS-60 unit (Bio-Logic) and the data were collected through a TIDAS diode array (J & M), with wavelength range between 300 and 1100 nm, all connected to a computer. The standard cuvette had an observation path length of 1 cm or 0.1 cm. For these experiments the dead time of each shot was previously determined to be 5.6 ms with a 8 mL s−1 flow rate.Semiempirical calculations
Geometries were optimized by AM1 at the UHF level.22Acknowledgements
Financial Support by FCT (project POCI/QUI/57735/2004) is acknowledged. FF is grateful for PhD grant POCTI/SFRH/BD/18905/2004.References
- F. Pina, M. Maestri and V. Balzani, Photochromic systems based on synthetic flavylium compounds and their potential use as molecular-level memory devices,Handbook of Photochemistry and Photobiology Vol. 3, ASP, Los Angeles, 2003, ch. 9, pp. 411–449 Search PubMed.
-
(a) R. Brouillard and J.-E. Dubois, J. Am. Chem. Soc., 1977, 99, 1359–1364 CrossRef CAS
 ;
(b) R. Brouillard and B. Delaporte, J. Am. Chem. Soc., 1977, 99, 8461–8468 CrossRef CAS ;
(c) R. Brouillard, B. Delaporte and J.-E. Dubois, J. Am. Chem. Soc., 1978, 100, 6202–6205 CrossRef CAS .
;
(b) R. Brouillard and B. Delaporte, J. Am. Chem. Soc., 1977, 99, 8461–8468 CrossRef CAS ;
(c) R. Brouillard, B. Delaporte and J.-E. Dubois, J. Am. Chem. Soc., 1978, 100, 6202–6205 CrossRef CAS . -
(a) R. A. McClelland and S. Gedge, J. Am. Chem. Soc., 1980, 102, 5838–5848 CrossRef CAS ;
(b) R. A. McClelland and G. H. McGall, J. Org. Chem., 1982, 47, 3730–3736 CrossRef CAS ;
(c) D. B. Devine and R. A. McClelland, J. Org. Chem., 1985, 50, 5656–5660 CrossRef CAS .
- P. Figueiredo, J. C. Lima, H. Santos, M. C. Wigand, R. Brouillard, A. L. Maçanita and F. Pina, J. Am. Chem. Soc., 1994, 116, 1249–1254 CrossRef CAS .
-
(a) F. Pina, L. Benedito, M. J. Melo, A. J. Parola and M. A. Bernardo, J. Chem. Soc., Faraday Trans., 1996, 92, 1693–1699 RSC ;
(b) F. Pina, M. J. Melo, M. Maestri, R. Ballardini and V. Balzani, J. Am. Chem. Soc., 1997, 119, 5556–5561 CrossRef CAS ;
(c) F. Pina, A. Roque, M. J. Melo, M. Maestri, L. Belladelli and V. Balzani, Chem.–Eur. J., 1998, 4, 1184–1191 CrossRef CAS ;
(d) F. Pina, M. J. Melo, A. J. Parola, M. Maestri and V. Balzani, Chem.–Eur. J., 1998, 4, 2001–2007 CrossRef CAS ;
(e) F. Pina, M. Maestri and V. Balzani, Chem. Commun., 1999, 107–114 RSC ;
(f) A. Roque, C. Lodeiro, F. Pina, M. Maestri, R. Ballardini and V. Balzani, Eur. J. Org. Chem., 2002, 2669–2709 ;
(g) M. C. Moncada, A. J. Parola, C. Lodeiro, F. Pina, M. Maestri and V. Balzani, Chem.–Eur. J., 2004, 10, 1519–1526 CrossRef CAS ;
(h) F. Pina, J. C. Lima, A. J. Parola and C. A. M. Afonso, Angew. Chem., Int. Ed., 2004, 43, 1525–1527 CrossRef CAS .
- H. Lietz, G. Haucke, P. Czerney and B. John, J. Prakt. Chem., 1996, 338, 725–730 CrossRef CAS .
- D. Amić, D. Davidović-Amić and D. Bešlo, J. Chem. Inf. Comput. Sci., 1999, 39, 967–973 CrossRef CAS .
- U.-W. Grummt and P. Czerney, J. Phys. Org. Chem., 2002, 15, 385–391 CrossRef CAS .
- H. Wünscher, G. Haucke, P. Czerney and U. Kurzer, J. Photochem. Photobiol., A, 2002, 151, 75–82 CrossRef CAS .
- F. Pina, J. Chem. Soc., Faraday Trans., 1998, 94, 2109–2116 RSC .
- F. Pina, M. J. Melo, M. Maestri, P. Passaniti, N. Camaioni and V. Balzani, Eur. J. Org. Chem., 1999, 3199–3207 CrossRef CAS .
- A. Roque, C. Lodeiro, F. Pina, M. Maestri, S. Dumas, P. Passaniti and V. Balzani, J. Am. Chem.
Soc., 2003, 125, 987–994 CrossRef CAS .
- M. C. Moncada, F. Pina, A. Roque, A. J. Parola, M. Maestri and V. Balzani, Eur. J. Org. Chem., 2004, 304–312 CrossRef CAS .
- M. C. Moncada, D. Fernández, J. C. Lima, A. J. Parola, C. Lodeiro, F. Folgosa, M. J. Melo and F. Pina, Org. Biomol. Chem., 2004, 2, 2802–2808 RSC .
- F. Galindo, J. C. Lima, S. V. Luis, A. J. Parola and F. Pina, Adv. Funct. Mater., 2005, 15, 541–545 CrossRef CAS .
- L. Giestas, F. Folgosa, J. C. Lima, A. J. Parola and F. Pina, Eur. J. Org. Chem., 2005, 4187–4200 CrossRef CAS .
- A. Jimenez, C. Pinheiro, A. J. Parola, M. Maestri and F. Pina, unpublished results.
- M. N. Berberan-Santos and J. M. G. Martinho, J. Chem. Educ., 1990, 67, 375–379 CrossRef CAS .
- J. S. Melo and A. L. Maçanita, Chem. Phys. Lett., 1993, 204, 556–562 CrossRef .
- D. D. Pratt and R. Robinsom, J. Chem. Soc., Trans., 1922, 121, 1577–1585 RSC .
- Ch. Michaelidis and R. Wizinger, Helv. Chim. Acta, 1951, 34, 1761–1770 CAS .
- Hyperchem™, release 7.0 for Windows, Hypercube, Inc., Gainesville, FL, USA Search PubMed .
|
| This journal is © The Royal Society of Chemistry 2007 |
Click here to see how this site uses Cookies. View our privacy policy here.