Spiroketals via oxidative rearrangement of enol ethers†
David L.
Waller
,
Corey R. J.
Stephenson
and
Peter
Wipf
*
Department of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania, USA. E-mail: pwipf@pitt.edu; Fax: +1 412-624-0787; Tel: +1 412-624-8606
First published on 14th November 2006
Abstract
Oxidative rearrangement of cyclic enol ethers leads to α-alkoxyesters. In the presence of a neighboring spiroether, this approach provides a stereoselective access to spiroketals. A modified proposal for the biosynthesis of acutumine is presented.
As part of our research program on the synthesis of highly oxygenated spiroketal natural products and Trx inhibitors,1 we attempted to model Barton's proposed biosynthesis of the spiro-fused vinylogous ester moiety of acutumine (1, Scheme 1).2 In this proposal, Barton suggested spirodienone 2 as a possible biosynthetic branching point. Double epoxidation of 2 followed by a hydrolytic Favorskii-type rearrangement furnishes acid 4. Decarboxylation and epoxide opening affords allylic diol 5, which is only a single oxidation level apart from vinylogous ester 6.3
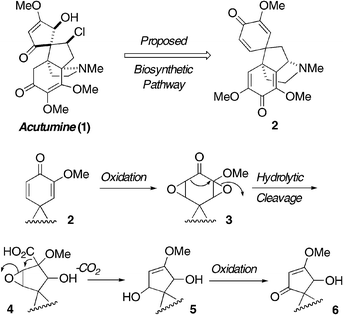 | ||
| Scheme 1 Barton's proposal for the biosynthesis of acutumine (1). | ||
Due to the ready access to dienones of type 2,4 this ring contraction provides an attractive, albeit hypothetical entry to acutuminealkaloid synthesis.5 Our first approach to investigate this proposal began with model dienone 7.6 Treatment with basic hydrogen peroxide provided monoepoxide 8 in 67% yield (Scheme 2). When 8 was treated with 3 equivalents of mCPBA buffered with Na2HPO4, we expected that the intermediate bis-epoxide would undergo Barton's proposed cascade reaction; however we isolated only racemic epoxylactone 9 in 80% yield as a single diastereomer, characterized by X-ray crystallography. We found that the actual diastereoselectivity of this reaction in the absence of Na2HPO4 was only 2 : 1; the crude product then readily equilibrated to a single diastereomer while washing the organic layer with aqueous NaHCO3.
 | ||
| Scheme 2 Reagents and conditions: (i) H2O2, K2CO3, THF, 40 °C, 6 h. (ii) m-chloroperbenzoic acid (3 eq.), Na2HPO4 (3 eq.), CH2Cl2, 18 h. | ||
While not entirely unprecedented,7 the epoxide rearrangement of 8 to 9 offers new and intriguing opportunities for natural product and diversity-oriented synthesis.8 In order to probe the mechanism and develop the scope of this transformation, we examined additional substrates, including five-and six-membered diosphenol ethers (10,91210), and the highly functionalized hydroxy enol ether 14. An epoxide (as in 8) was clearly not necessary: enone 10 underwent rearrangement to lactone 11 in 75% yield (Scheme 3). In contrast, the yield for the conversion of five-membered enone 12 to lactone 13 was only 35%. A carbonyl functionality was also not needed to facilitate the rearrangement; the epoxy alcohol 14, prepared by the addition of MeLi to ketone 8, succumbed to the oxidative rearrangement to give hemiacetal 15 in 20% yield as a racemic single diastereomer which was again characterized by X-ray crystallography.
 | ||
| Scheme 3 Reagents and conditions: (i) m-chloroperbenzoic acid (3 eq.), Na2HPO4 (3 eq.), CH2Cl2, 18 h. | ||
A proposal for the oxidative rearrangement of 10 is outlined in Scheme 4. The epoxy ether intermediate 16 opens to the alkoxycarbenium ion 17 in the presence of a proton donor. This species is then intercepted by another equivalent of peracid to give peroxy ketal 18. A methyl ether assisted acyl shift generates the seven-membered lactone 19 which can undergo a ring contraction to generate the product lactone ester 20, which is epimerizable at the α-position to the ester moiety, resulting in the thermodynamically more stable equatorial ester after basic workup and isolation. This sequence constitutes a net addition of two oxygen atoms concomitant with a skeletal rearrangement.7
 | ||
| Scheme 4 Proposed mechanism for oxidative rearrangement. | ||
Even though the product forms in low yield, we were particularly interested in the possible use of highly functionalized substrates such as 14 in the oxidative enol ether rearrangement. Replacement of the tertiary hydroxy group with a cyclic ether would establish a novel access to spiroacetals, which are common features in biologically active natural products.11,12 A procedure developed by Paquette et al. provided rapid access to the desired α-carbonyl functionalized cyclic ethers.13 Addition of 5-lithio-2,3-dihydrofuran, to cyclopentanone furnished an intermediate tertiary allylic alcohol which underwent an acid catalyzed pinacol rearrangement to give spirocyclic ketone 2113 in 65% overall yield (Scheme 5). While hard electrophiles such as Meerwein's reagent failed to give satisfactory yields of O-alkylation, generation of the enolate of 21 with KHMDS, followed by addition of DMF and dimethyl sulfate furnished the methyl enol ether 22 in 89% yield. When 22 was submitted to the oxidative rearrangement with mCPBA, the volatile spiroketal 23 was obtained as the sole product in 52% yield.
 | ||
| Scheme 5 Reagents and conditions: (i) 5-lithio-2,3-dihydrofuran,THF, –78 °C, 12 h. (ii) Dowex 50X, CH2Cl2, 18 h. (iii) KHMDS, Me2SO4, THF–DMF (4 : 1), –78 °C, 4 h. (iv) m-chloroperbenzoic acid (3 eq.), Na2HPO4 (3 eq.), CH2Cl2, 18 h. | ||
For a further expansion of this methodology, we prepared methyl enol ethers 24, 2614 and androsterone-based 2814 in a fashion analogous to 22.15 When 24 was submitted to the rearrangement, the volatile spiroketal 25 was isolated in 48% yield as a modest 2 : 1 mixture of diastereomers (Scheme 6). The tricyclic methyl enol ether 26 smoothly underwent the rearrangement in 53% yield, and product 27 was isolated as a single diastereomer. Additionally, 28 was also converted to pentacyclic spiroketal 29 as a single diastereomer in 76% yield.16
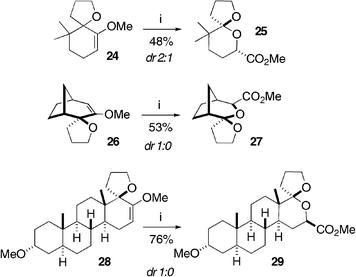 | ||
| Scheme 6 Reagents and conditions: (i) m-chloroperbenzoic acid (3 eq.), Na2HPO4 (3 eq.), CH2Cl2, 18 h. | ||
In conclusion, we have demonstrated a new and efficient oxidative rearrangement of alkyl enol ethers to lactone and spiroketal esters. Our method allows a rapid access to these common structural subunits of natural products. We are currently exploring additional applications of this process towards biologically active molecules. This investigation was inspired by Barton's proposal for the biosynthesis of acutumine, but we have not been able to garner any experimental support for the dienone diepoxide rearrangement shown in Scheme 1. While an enzymatic pathway could easily take a different course, it appears that, chemically, α-epoxy ethers of type 3 prefer alternative rearrangements to a migratory ring contraction. In fact, the recent isolation of acutudaurin,17 a possible precursor of acutumine-type natural products, supports a modified biosynthetic pathway (Scheme 7).
 | ||
| Scheme 7 Alternative proposal for the biosynthesis of acutumine (1). | ||
The tricarbonyl tyrosine dimer 30 can be envisioned as a direct precursor of 1 after oxidation and benzilic acid rearrangement (31 → 32), followed by decarboxylation to give the cyclopentanone subunit 6. This pathway is supported by experimental observations in the literature,18 and the oxygenation pattern on spirocycle 31 is in good agreement with the structure of acutudaurin.
Acknowledgements
This work has been supported the NIH/NIGMS CMLD program (GM067082). The authors thank Mr Alvarro Carraro and Dr Hidenori Takahashi (University of Pittsburgh) for preliminary synthetic studies, Dr Katsuhide Matoba (Otsuka Pharmaceutical Co. Ltd) for stimulating discussions, and Dr Steve Geib (University of Pittsburgh) for X-ray analysis.References
- For recent examples, see: (a) G. Powis, P. Wipf, S. M. Lynch, A. Birmingham and D. L. Kirkpatrick, Mol. Cancer Ther., 2006, 5, 630 Search PubMed; (b) P. Wipf, S. M. Lynch, G. Powis, A. Birmingham and E. E. Englund, Org. Biomol. Chem., 2005, 3, 3880 RSC; (c) P. Wipf, S. M. Lynch, A. Birmingham, G. Tamayo, A. Jimenez, N. Campos and G. Powis, Org. Biomol. Chem., 2004, 2, 1651 RSC; (d) P. Wipf and J.-K. Jung, J. Org. Chem., 2000, 65, 6319 CrossRef CAS.
- D. H. R. Barton, A. J. Kirby and G. W. Kirby, J. Chem. Soc. C, 1968, 929 RSC.
- The biosynthetic pathways for acutumine have been investigated: H. A. A. Babiker, Y. Sugimoto, T. Saisho and S. Inanaga, Phytochemistry, 1999, 50, 775 Search PubMed; H. A. A. Babiker, Y. Sugimoto, T. Saisho, S. Inanaga, M. Hashimoto and A. Isogai, A., Biosci., Biotechnol., Biochem., 1999, 63, 515 CrossRef CAS; Y. Sugimoto, S. Uchida, S. Inanaga, Y. Kimura, M. Hashimoto and A. Isogai, Biosci., Biotechnol., Biochem., 1996, 60, 503 CrossRef CAS.
- P. Wipf and Y. Kim, J. Am. Chem. Soc., 1994, 116, 11678 CrossRef CAS.
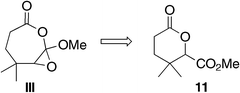
- Barton's proposal has been investigated before, although on a simpler model system: K. Matoba, N. Karibe and T. Yamazaki, Chem. Pharm. Bull., 1984, 32, 2639 Search PubMed . This group assigned an enone oxidation product as a Baeyer–Villiger derived lactone. Based on our investigation presented herein, we believe that their assignment of III should be revised to 11
. - K. Matoba, N. Karibe and T. Yamazaki, Chem. Pharm. Bull., 1982, 30, 3906 CAS.
- To the best of our knowledge, this net transformation has been observed only twice; in both cases upon treatment of enones with basic hydrogen peroxide for extended periods of time: K. L. Bhat and G. Trivedi, Synth. Commun., 1982, 12, 585 Search PubMed; M. Kocor, A. Kurek and J. Dabrowski, Tetrahedron, 1969, 25, 4257 CrossRef CAS.
- For lead references on the concept of diversity-oriented synthesis, see: (a) D. S. Tan, Nat. Chem. Biol., 2005, 1, 74 CrossRef CAS; (b) P. Wipf, C. R. J. Stephenson and M. A. A. Walczak, Org. Lett., 2004, 6, 3009 CrossRef CAS; (c) P. Wipf, S. Werner, G. H. C. Woo, C. R. J. Stephenson, M. A. A. Walczak, C. M. Coleman and L. A. Twining, Tetrahedron, 2005, 61, 11488 CrossRef CAS.
- (a) E. Wenkert, N. F. Golob, S. S. Sathe and R. A. J. Smith, Synth. Commun., 1973, 3, 205 CrossRef CAS; (b) G. F. Hennion and F. P. Kupiecki, J. Org. Chem., 1953, 18, 1601 CrossRef CAS.
- B.-M. Kwon and C. S. Foote, J. Org. Chem., 1989, 54, 3878 CrossRef CAS.
- Reviews: (a) K. T. Mead and B. N. Brewer, Curr. Org. Chem., 2003, 7, 227 CrossRef CAS; (b) M. A. Brimble and F. A. Fares, Tetrahedron, 1999, 55, 7661 CrossRef CAS; (c) S. Rodriguez and P. Wipf, Synthesis, 2004, 2767 CAS.
- For recent synthetic work, see: (a) J. Hao, F. Matsuura, Y. Kishi, M. Kita, D. Uemura, N. Asai and T. Iwashita, J. Am. Chem. Soc., 2006, 128, 7742 CrossRef CAS; (b) C. Wang and C. J. Forsyth, Org. Lett., 2006, 8, 2997 CrossRef CAS; (c) K. Meilert and M. A. Brimble, Org. Biomol. Chem., 2006, 4, 2184 RSC; (d) V. Rauhala, K. Nattinen, K. Rissanen and A. M. P. Koskinen, Eur. J. Org. Chem., 2005, 4119. I; (e) I. Paterson, D. Gottschling and D. Menche, Chem. Commun., 2005, 3568 RSC; (f) P. Wipf and T. D. Hopkins, Chem. Commun., 2005, 3421 RSC.
- L. A. Paquette, J. T. Negri and R. D. Rogers, J. Org. Chem., 1992, 57, 3947 CrossRef CAS.
- The assignment of the configuration of the tetrahydrofuran ring was based on previous studies conducted on these acid-mediated ring expansions: L. A. Paquette, J. F. P. Andrews, C. Vanucci, D. E. Lawhorn, J. T. Negri and R. D. Rogers, J. Org. Chem., 1992, 57, 3956 Search PubMed.
- See supporting information for experimental details.
- The configurations of 25 and 27 were assigned in analogy to 9 and 15, which were determined by X-ray analysis. CCDC reference numbers 624751 and 624752. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b612992g. Further support for this assignment was obtained by 1H NMR coupling constant analysis of the hydrogen α to the methyl ester. Compound 26 is the methyl enol ether of a known compound as noted in ref. 14.
- T. Furumoto and Y. Sugimoto, Planta Med., 2001, 67, 194 CrossRef CAS.
- (a) V. Georgian and N. Kundu, Tetrahedron, 1963, 19, 1037 CrossRef CAS; (b) N. Greenhalgh, J. Chem. Soc., 1954, 4699 Search PubMed; (c) H. Liu, D. R. Siegel and S. J. Danishefsky, Org. Lett., 2006, 8, 423 CrossRef CAS; (d) R. Holzel, A. P. Leftwick and B. C. L. Weedon, J. Chem. Soc. D, 1969, 128 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data for new compounds; X-ray data for compounds 9 and 15. See DOI: 10.1039/b612992g |
| This journal is © The Royal Society of Chemistry 2007 |