Synthesis of supramolecular fullerene–porphyrin–Cu(phen)2–ferrocene architectures. A heteroleptic approach towards tetrads†
Michael
Schmittel
*a,
Ravuri S. K.
Kishore
a and
Jan W.
Bats
b
aCenter of Micro and Nanochemistry and Engineering, Organische Chemie I, Universität Siegen, Adolf-Reichwein-Str., D-57068, Siegen, Germany. E-mail: schmittel@chemie.uni-siegen.de; Fax: +49 271 740 3270
bInstitut für Organische Chemie und Chemische Biologie, Johann Wolfgang Goethe-Universität, Marie-Curie-Strasse 11, D-60439, Frankfurt, Germany
First published on 16th November 2006
Abstract
Four supramolecular fullerene–porphyrin–Cu(phen)2–ferrocene architectures were accessed by a twofold coordination strategy. At first, the phenanthroline-linked zinc porphyrins 1–4, conceived as supramolecular synthons, were combined with a ferrocene module, 3,8-(diferrocenylethynyl)phenanthroline (5), by a Cu(I)-mediated heteroleptic bisphenanthroline complexation (HETPHEN) protocol to furnish the porphyrin–Cu(phen)2–ferrocene aggregates 6–9. Subsequently, the fullerene module 10 was incorporated by axial pyridyl coordination to the zinc porphyrin, affording 11–14. Their suitability as tetrads was interrogated using electrochemical and photophysical data.
Introduction
The conversion of solar into chemical energy is probably the most important and consequential step occurring in the photosynthetic reaction centre in plants.1 In purple photosynthetic bacteria for instance, efficient light harvesting and excitation energy transfer (EET) funnels solar energy from the photosystems LH2 and LH1 to the special pair bacteriochlorophyll dimer (Bchl2).1 The subsequent cascade of very fast electron transfer (eT) steps produces a charge-separated (CS) state Bchl2˙+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Qa˙− (quinone) over a fairly large distance.2 Charge recombination (CR) is effectively avoided until the hole and electron are separated by a membrane.
Qa˙− (quinone) over a fairly large distance.2 Charge recombination (CR) is effectively avoided until the hole and electron are separated by a membrane.
Major efforts have been made in past decades towards mimicking this process and in developing artificial photosynthetic systems.3,4 An important class of compounds showing long-lived CS states are the fullerene-porphyrin-ferrocene triads, tetrads and pentads.5 In particular, highly efficient photosynthetic electron transfer has been realised by Fukuzumi, Guldi, Imahori, Ito and colleagues6 in ferrocene–zinc porphyrin–C60 triads, in which the relatively long-lived CS state (up to 16 µs) could be produced with an extremely high quantum yield (nearly unity). They also reported a ferrocene–zinc porphyrin trimer–fullerene pentad,7 which exhibited not only the longest lifetime (0.53 s in PhCN at 163 K) of a CS state, but also an extremely high CS efficiency (83%), comparable to the eT properties of bacteriochlorophyll dimer radical cation (Bchl2˙+)–secondary quinone radical anion (QB˙−) ion pair in the bacterial photosynthetic reaction centre.
However, while in the natural photosynthetic reaction centre the eT process occurs along a supramolecular relay system in a protein matrix, most of the above-mentioned reports represent covalent systems. Construction of well defined structures bearing three or more different functional photo/redox active units is tedious in a dynamic supramolecular regime.4a,d,8 It is hence not surprising that very few reports of supramolecular triads,9 and none of tetrads, are known. Invariably, in the fullerene–porphyrin–ferrocene triads known so far9 there is only one non-covalent binding motif present, and either the fullerene or the ferrocene is covalently bound to the porphyrin. A supramolecular triad/tetrad where both the ferrocene and the fullerene subunits are bound solely by non-covalent interactions has been difficult to access. In the present study, we demonstrate how a combination of two orthogonal binding modes – involving (i) Cu(I)-mediated heteroleptic bis-phenanthroline complexation and (ii) axial binding of pyridine to zinc porphyrin – is used effectively to obtain four supramolecular fullerene–porphyrin–Cu(phen)2–ferrocene aggregates, as a proof of principle for the heteroleptic construction of tetrads. The Cu(phen)2 complex, apart from playing a structural role as a non-covalent binding unit, is known to efficiently mediate electron transfer between Zn(II) and Au(III) porphyrins, as reported by Sauvage,10 and is also known to be involved in photoinduced eT processes with the C60 moiety.11
Results and discussion
The strategy to the supramolecular aggregates involves three steps: (i) synthesis of phenanthroline-linked porphyrins 1–4; (ii) Cu(I)-driven heteroleptic bis-phenanthroline complexation of 1–4 with bis(ferrocenylethynyl)phenanthroline 5 to afford the Cu(I)-mediated supramolecular porphyrin–Cu(phen)2–ferrocene aggregates 6–9; and (iii) axial coordination of pyridyl fullerene 10 to 6–9 to furnish the supramolecular fullerene–porphyrin–ferrocene systems 11–14.Synthesis of phenanthroline-appended porphyrins 1–4
Our strategy towards the synthesis of 1–4 was to attach phenanthroline12Phen1 to the iodophenyl groups in porphyrins P1–P4via a Sonogashira cross-coupling. Compounds 1 and 4 have already been reported by us elsewhere.13In order to access the required phenanthroline–porphyrin hybrids 1–4, the meso-iodophenyl zinc-porphyrins P1–P4 were first synthesised based on strategies outlined by Lindsey et al.14 A subsequent Sonogashira reaction using Pd2(dba)3/AsPPh3 in benzene or pyridine with triethylamine as a base allowed for the cross-coupling between Phen1 and P1–P4, to furnish 1–4 (Scheme 1). Purification of 1–4 was realised by chromatography on silica-gel followed by size-exclusion chromatography on bio-beads in toluene.
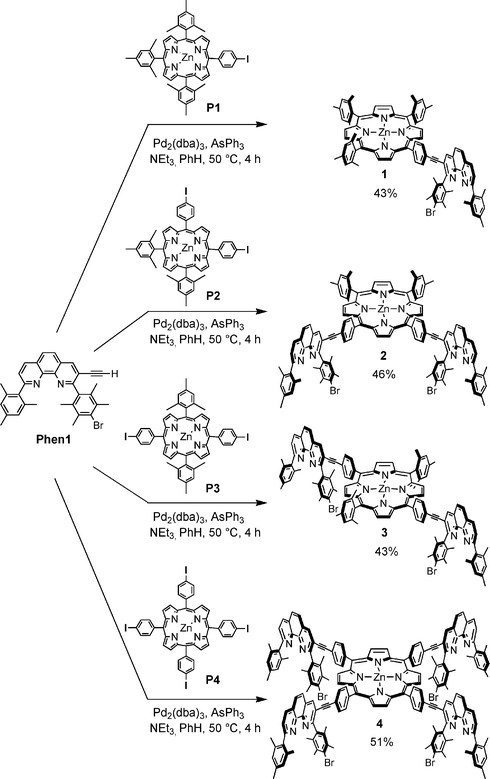 | ||
| Scheme 1 Synthesis of phenanthroline–porphyrin hybrids 1–4. | ||
Table S1 (see ESI†) displays the various NMR shifts of 1–4 measured in CD2Cl2. It was noticed that the Hβ protons of the pyrrole units of the porphyrin rings were split according to the symmetry of the ligands. The distinct attachment in the meso position of the porphyrin did, however, not entail tangible NMR shift differences of the phenanthroline protons. The sole difference noted was an upfield shift for the 4,7- and 5,6-protons on the phenanthroline unit of 4, compared to their counterparts in 1, 2, and 3.
Absorption spectroscopy (Table 1) showed an increasing bathochromic shift of the Soret band from 1 to 4 relative to zinc 5,10,15,20-tetraphenylporphyrin (ZnTPP), i.e. with the growing number of phenanthrolines at the periphery of the zinc porphyrin core (Fig. 1). Also noticed was the predicted increase in the absorption coefficient of the phenanthroline chromophore in 1–4 in the 250–350 nm region. The ligands displayed strong fluorescence when excited at the Soret band.
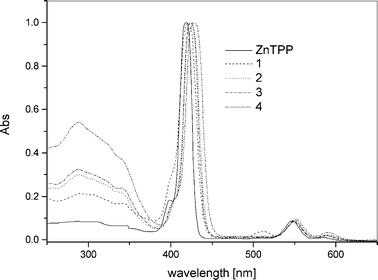 | ||
| Fig. 1 Absorption spectra of 1–4 measured in CH2Cl2 normalised at the Soret band. | ||
Formation of porphyrin–Cu(phen)2–ferrocene aggregates by the HETPHEN complexation strategy
Ligands 1–4 are supramolecular building blocks, which can be utilised for the construction of porphyrin–Cu(phen)2–ferrocene systems by a metal-driven heteroleptic complexation. As a proof of concept, we have recently incorporated phenanthrolinyl porphyrin 1 into a multicomponent self-assembled stack.13b Along the same lines, the tetrakis(bis-heteroleptic) supramolecular phenanthroline complex 9 was realised starting from porphyrin 4 through a Cu(I)-mediated approach.13a Accordingly, the supramolecular aggregates 6–8 (Chart 1) were prepared based on the HETPHEN15 approach. For this purpose, compound 5 was synthesised by a Sonogashira reaction of ferrocenylacetylene with 3,8-dibromophenanthroline using [Pd(PPh3)2]Cl2/CuI and NHiPr2 (Scheme 2). Single crystals of 5 were obtained by slow diffusion of hexane into a concentrated solution of 5 in chloroform. Fig. 2 displays the crystal structure of 5.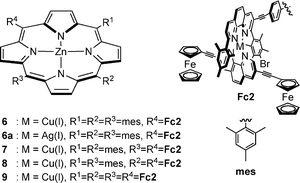 | ||
| Chart 1 Chemical representation of triads 6–9 and 6a. | ||
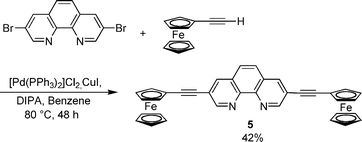 | ||
| Scheme 2 Synthesis of diferrocenylethynylphenanthroline 5. | ||
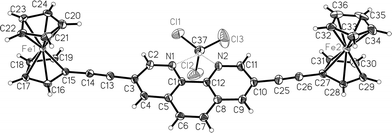 | ||
| Fig. 2 Crystal structure of 5 (the non-H atoms are shown with 50% probability ellipsoids). | ||
Formation of supramolecular fullerene–porphyrin–Cu(phen)2–ferrocene systems
Reaction of 6–9 with 10 in CH2Cl2–CS2 (1 : 1) afforded 11–14 (Scheme 3, Chart 2). Their formation was readily detected by UV-vis spectroscopy, because the Soret band of 11–14 displayed a characteristic bathochromic shift from 420 nm to 430 nm. In addition, characteristic shifts of various signals in the 1H NMR were noticed. Notably, the Hβ pyrrole signals of the porphyrin were found to shift by ∼0.1 ppm upfield in all complexes. The signal for the 3″,5″-mesitylene protons (C6H2Me3) experienced a downfield shift from δ 6.14 in 6–9 to δ 6.21 in 11–14. (Table S2†). Likewise, the signals corresponding to the ferrocene protons were found shifted upfield by ∼0.1 ppm. This could arise due to the proximity of the ferrocene to the fullerene subunit.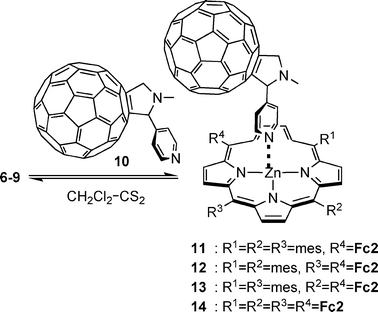 | ||
| Scheme 3 Reaction leading to formation of 11–14. | ||
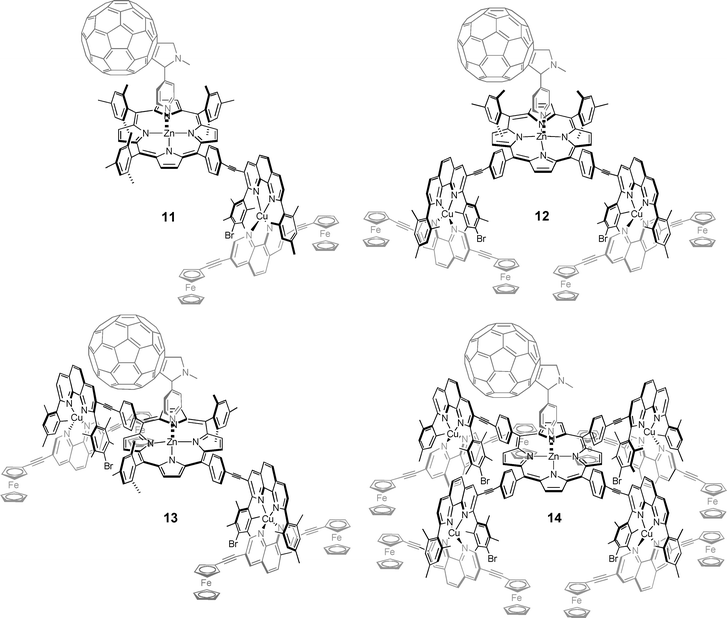 | ||
| Chart 2 Chemical representation of tetrads 11–14. | ||
The 1H NMR spectra of 6 and 11 are shown in Fig. 3. A distinct upfield shift of the 3,5-pyridyl protons of 10 from δ 7.76 ppm (as free residue) to a broad singlet at δ 6.44 (in 11) was noticed. The signals of the protons from the pyrrolidine ring fused to C60, at δ 4.67 and 3.92, also experienced an upfield shift from δ 5.02 and 4.32 in 10. Similar shifts were found in 12, 13 and 14. Due to the density of methyl signals, the 2,6-pyridyl protons, known to shift drastically upfield to the region δ 0.5–2.0, were not readily discernible.
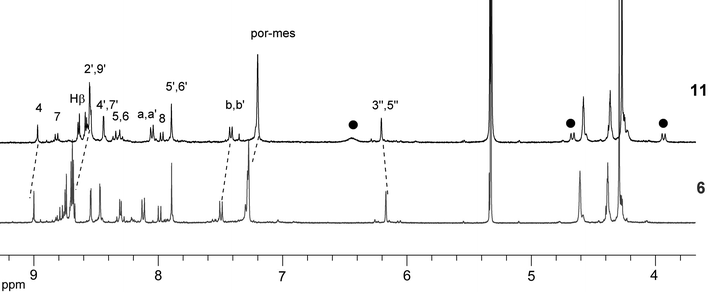 | ||
| Fig. 3 Aromatic region in the 1H NMR spectra of 6 and 11. The solid black circles represent the shifts of the 3,5-H and the pyrrolidine protons. | ||
The sharp signals in the NMR spectra suggest that there is free rotation about the various axes present in 11–14. In addition, no broadening of any signals in 11–14 was observed compared to 6–9. Presence of a single set of ferrocene signals was noticed in 11, 12 and 14, implying equal interaction of the fullerene with all the ferrocene protons.
ESI-MS characterisation16 revealed molecular ion signals for 11–14, not unexpectedly accompanied by signals from 6–9. Within the range of the instrument, isotopic splitting patterns were obtained for 12–14, matching with those of the simulated spectra.
UV-Visible absorption titrations of 6–9 with 10 in dichloroethane at 298 K exhibited spectral changes characteristic of axial coordination of a pyridine ligand to a zinc porphyrin.17 The formation constants K for the porphyrin–fullerene conjugates could be determined from the spectral data by SPECFIT.18 The association constants are listed in Table 2. Notably, the association constant for formation of 11–14 (from 6–9 and 10) is higher by half an order of magnitude than that of ZnTPP and 10.
| K/M−1 | logβ | E 0–0/eV | reference | |
|---|---|---|---|---|
| a Measured in ortho-dichlorobenzene at 298 K. b Measured in DCE at 298 K. | ||||
| ZnTPP a | 7.2 × 103 | 3.86 (± 0.38) | — | 5d |
| 11 b | 2.1 × 104 | 4.34 (± 0.05) | 1.96 | this work |
| 12 b | 2.3 × 104 | 4.36 (± 0.02) | 1.96 | this work |
| 13 b | 1.6 × 104 | 4.19 (± 0.07) | 1.96 | this work |
| 14 b | 2.6 × 104 | 4.42 (± 0.03) | 1.96 | this work |
The molecular structure of aggregate 11 calculated at the PM3 level in SPARTAN19 showed an edge-to-edge distance of 4.1 Å between the ferrocene and the C60 unit, indicative of an almost complete van der Waal's contact between the ferrocene and the fullerene unit, as reported earlier by D'Souza9aet al. for other systems. This may explain the higher association constants displayed by 11–14 and the upfield-shifted signals of the ferrocene protons in the 1H NMR.
Compounds 11–14 are supramolecular aggregates built purely by a supramolecular assembly protocol by applying two dynamic non-interfering binding modes. Certainly, the heteroleptic Cu(I)–bisphenanthroline complex is a dynamic yet robust (logβ ∼9.7) supramolecular motif, as we have recently shown by demonstrating the exchange of metal ions (Cu(I) vs. Ag(I)), as well as of ligands.13 The latter was found to be completely arrested only at −60 °C. Axial binding of the pyridyl fullerene to zinc porphyrin (logK ∼4.3) is comparatively more dynamic. This is evidenced by broadened signals of the pyridyl protons in the 1H NMR of 11–14 at room temperature. However, an earlier study by Imamura20 stated that below −40 °C the dynamic nature of this system was arrested, as seen from the sharpening of the signals of the pyridyl protons in the 1H NMR.
Electrochemical studies
The electrochemical properties of complexes 11–14 were investigated along with those of 1–9 (Table 3 and Table S3†). The redox potentials were measured by differential pulse voltammetry (DPV) and cyclic voltammetry in dichloroethane. The intactness of the supramolecule in the presence of electrolyte NBu4PF6 under the DPV and CV conditions was proven by absorption spectroscopy.| E 1/2 vs. Fc a | HOMO–LUMO/eVb | ||||||
|---|---|---|---|---|---|---|---|
| P+/2+ | P0/+, Cu+/2+ | Fc0/+ | C600/− | C60−/2− | P0/− | ||
| a Determined by DPV in dichloroethane with NBu4PF6 as electrolyte, and measured against an Ag wire as a quasi-reference electrode and dimethylferrocene as internal standard at 100 mV s−1. Potentials are referenced against the more common ferrocene redox couple. b Determined from the redox potentials Fc0/+ − C600/−. c Deconvolution allowed to separate the overlapping waves of the P0/+ and Cu+/2+ redox processes. d Obtained by deconvolution. | |||||||
| 6 | 0.74 | 0.41, 0.41 | 0.21 | — | — | −1.91 | — |
| 6a | 0.74 | 0.41 | 0.21 | — | — | — | — |
| 7 | 0.77 | 0.41, 0.41 | 0.20 | — | — | −1.86 | — |
| 8 | 0.74 | 0.41, 0.41 | 0.21 | — | — | −1.86 | — |
| 9 | 0.77 | 0.41, 0.41 | 0.20 | — | — | −1.84 | — |
| 11 | 0.85 | 0.37, 0.30c | 0.21d | −1.06 | −1.47 | −1.98 | 1.27 |
| 12 | 0.83 | 0.42, 0.36c | 0.21 | −1.04 | −1.44 | −1.88 | 1.25 |
| 13 | 0.77 | 0.39, 0.33c | 0.20 | −1.08 | −1.50 | −1.88 | 1.28 |
| 14 | 0.81 | 0.40, 0.28c | 0.19 | −1.04 | −1.44 | −1.85 | 1.23 |
Redox data of 6–9 (Table 3) were measured in dichloroethane by differential pulse voltammetry (DPV). It was observed that the redox potentials of P+/2+, P0/+, Cu+/2+ and ferrocene (Fc0/+) in 6–9 were not influenced by the number of phenanthrolines at the porphyrin periphery. In contrast, the porphyrin reduction potential (P0/−) exhibited an anodic shift of ∼70 mV while going from 6 to 9 (Table 3). The number of electrons transferred in the individual redox steps, as analysed from the integrated areas of the DPV curves, indicated that the first oxidation of the porphyrin (P0/+) and that of Cu+/2+ overlapped at E = 0.41 VFc. This assignment was confirmed by preparing 6a, the silver analog of 6, containing an Ag(I) instead of the Cu(I) phenanthroline complex. As for 6, the porphyrin E1/2 (P0/+) in 6a was located at E = 0.41 VFc.
Table 3 shows the redox potentials of 11–14. On the basis of two criteria (peak separation Epa − Epc and ipa/ipc = 1) the redox processes were found to be reversible. In contrast to systems 6–9, the redox waves for Cu+/2+ and P0/+ in 11–14 only partly overlapped, so that we could employ a peak deconvolution with PEAKFIT for their analysis. The E1/2 (P0/+) of the porphyrin unit in 11–14 exhibited small, but uncharacteristic shifts when compared to 6–9, while the reduction potential E1/2 (P0/−) revealed cathodic shifts of 0–70 mV. The porphyrin potentials E1/2 (P0/+) of 11–14 are higher than that of ZnTPP·10 by 200 mV, mostly due to the electron-withdrawing effect of the Cu+/2+ centres and to a lesser extent because of the charged Fc˙+ groups.
Due to the spatial arrangement and electrochemical data of 11–14 we expect that upon photochemical excitation of the porphyrin unit a PET process will generate C60˙−–Por˙+–Cu(phen)2+–Fc, which will undergo further CS to C60˙−–Por–Cu(phen)22+–Fc and finally to C60˙−–Por–Cu(phen)2+–Fc˙+. Hence, the present system should operate as a A–D–D–D tetrad, with the CS from C60˙−–Por˙+–Cu(phen)2+–Fc to C60˙−–Por–Cu(phen)22+–Fc having almost no driving force. A caveat of the present discussion is that the redox data in Table 3 do not precisely reflect the redox situation in the photochemically triggered tetrads. Obviously, the PET process will be initiated with the ferrocene units being uncharged and the copper being singly-charged, while the data for P0/+ recorded in Table 3 are influenced by the oxidised ferrocene and the doubly-charged copper units.
Conclusions
We have successfully established a protocol to obtain dynamic supramolecular aggregates based on two orthogonal coordinative binding motifs. The successful introduction of a Cu(phen)2 redox unit into the tetrad system should facilitate stepwise charge separation, as has been noticed previously with other supramolecular CS systems.10,11,21 Moreover, the present self-assembly approach leaves a lot of room for further fine-tuning of the electronic properties of triads and tetrads, e.g. using other spacers in between the coordination motifs, installing different terminal redox relays and even modulating the redox properties of the intermediate copper–bisphenanthroline linkage. Further investigations on these supramolecular tetrads are underway.Experimental
Synthesis
Starting materials Phen1 and P1–4 were synthesised according to known procedures.12,14 Ligands 1,13b4,13a513a and 109 were prepared according to previous reports. 1H and 13C NMR were measured on a Bruker Avance 400. ESI-MS were measured on a ThermoQuest LCQ Deca instrument. Typically, 25 scans were accumulated for one spectrum. All complexes were characterised by 1H NMR, 13C NMR, and elemental analysis. Cyclic voltammetry and differential pulse voltammetry were measured on a Parstat 2273 potentiostat.Using porphyrin P1 (138 mg, 148 µmol), Phen1 (79 mg, 148 µmol), Pd2(dba)3 (10 mg, 1.5 µmol), AsPh3 (45 mg, 148 µmol). Yield of 1: 85 mg (43%, 63 µmol); δH (400 MHz, CD2Cl2) 8.84 (d, J = 4 Hz, 2 H, 2,18-Hβ), 8.75 (d, J = 4 Hz, 2 H, 3,17-Hβ), 8.70 (s, 4 H, 7,8,12,13-Hβ), 8.66 (s, 1 H, 4-H), 8.36 (d, J = 8 Hz, 1 H, 7-H), 8.16 (d, J = 8 Hz, 2 H, Ar-Hb,Hb′), 7.97 (br s, 2 H, 5,6-H), 7.61 (d, J = 8 Hz, 1 H, 8-H), 7.51 (d, J = 8 Hz, 2 H, Ar-Ha,Ha′), 7.29 (s, 6 H, 3‴,5‴-H), 6.98 (s, 2 H, 3″,5″-H), 2.62 (s, 9 H, 8‴,8⁗-H ), 2.55 (s, 6 H, 8′,9′-H), 2.35 (s, 3 H, 8″H), 2.14 (s, 6 H, 7″,9″-H), 2.07 (s, 6 H, 7′,10′-H), 1.84 (s, 6 H, 7⁗,9⁗-H), 1.83 (s, 6 H, 7‴,9‴-H); δC (100 MHz, CDCl3) 163.0, 161.1, 150.5, 150.3, 150.2, 149.7, 146.6, 145.5, 144.9, 142.9, 140.7, 139.7, 138.9, 138.4, 138.0, 137.7, 136.5, 135.1, 134.9, 134.2, 133.8, 132.1, 131.5, 131.1, 130.9, 130.0, 129.7, 129.3, 128.9, 128.8, 128.7, 128.0, 127.6, 127.4, 127.1, 126.1, 125.7, 122.0, 120.7, 119.2, 119.1, 96.3, 88.1, 22.2, 22.1, 21.8, 21.5, 21.4, 20.7, 18.9; ESI-MS m/z (%): 1335.9 (100) [M + H]+; Found: C, 77.74; H, 5.68; N, 6.28. Calcd for C86H73BrN6Zn: C, 77.32; H, 5.51; N, 6.29%.
Using porphyrin P2 (190 mg, 187 µmol), Phen1 (200 mg, 375 µmol), Pd2(dba)3 (18 mg, 1.9 µmol), AsPh3 (57 mg, 187 µmol). Yield of 2: 157 mg (46%, 86 µmol); δH (400 MHz, CD2Cl2) 8.93 (s, 2 H, 2,3-Hβ), 8.88 (d, 2 H, J = 4 Hz, 7,18-Hβ), 8.80 (d, J = 4 Hz, 2 H, 8,17-Hβ), 8.75 (s, 2 H, 12,13-Hβ), 8.68 (s, 2 H, 4-H), 8.33 (d, J = 8 Hz, 2 H, 7-H), 8.17 (d, J = 8 Hz, 4 H, Ar-Hb,Hb′), 7.92 (m, 4 H, 5,6-H), 7.59 (d, J = 8 Hz, 2 H, 8-H), 7.55 (d, J = 8 Hz, 4 H, Ar-Ha,Ha′) 7.31 (s, 4 H, 3‴,5‴-H), 7.00 (s, 4 H, 3″,5″-H), 2.63 (s, 6 H, 8‴-H), 2.57 (s, 12 H, 8′,9′-H), 2.37 (s, 6 H, 8″-H), 2.17 (s, 12 H, 7″,9″-H), 2.09 (s, 12 H, 7′,10′-H), 1.85 (s, 12 H, 7‴,9‴-H); δC (100 MHz, CD2Cl2) 162.7, 160.9, 150.5, 150.2, 150.1, 149.9, 146.4, 145.3, 143.9, 139.8, 139.5, 139.2, 138.9, 138.4, 138.0, 137.9, 136.5, 136.1, 134.9, 134.3, 133.9, 132.3, 131.9, 131.6, 131.3, 130.0, 129.4, 128.6, 128.0, 128.0, 127.5, 127.4, 126.0, 125.2, 122.1, 120.3, 119.9, 119.7, 95.9, 87.9, 21.8, 21.5, 21.2, 21.2, 20.5, 18.7; ESI-MS m/z (%): 1825.8 (90) [M + H]+, 913.7 (100) [M + 2H]2+; Found: C, 76.48; H, 5.15; N, 5.84. Calcd for C116H94Br2N8Zn: C, 76.33; H, 5.19; N, 6.14%
Using porphyrin P3 (67 mg, 86 µmol), Phen1 (97 mg, 182 µmol), Pd2(dba)3 (21 mg, 2.2 µmol), AsPh3 (26 mg, 86 µmol). Yield of 3: 130 mg (43%, 37 µmol); δH (400 MHz, CD2Cl2–CD3OD) 8.77 (d, 4 H, J = 4 Hz, 2,8,12,18-Hβ), 8.67 (d, 4 H, J = 4 Hz, 3,7,13,17-Hβ), 8.65 (s, 2 H, 4-H), 8.36 (d, J = 8 Hz, 2 H, 7-H), 8.13 (d, J = 8 Hz, 4 H, Ar-Hb,Hb′), 7.96 (m, 4 H, 5,6-H), 7.58 (d, J = 8 Hz, 2 H, 8-H), 7.47 (d, J = 8 Hz, 4 H, Ar-Ha,Ha′), 7.27 (s, 4 H, 3‴,5‴-H), 6.96 (s, 4 H, 3″,5″-H), 2.60 (s, 6 H, 8‴-H), 2.53 (s, 12 H, 7‴,9‴-H), 2.33 (s, 6 H, 8″-H), 2.12 (s, 12 H, 7′,10′-H), 2.05 (s, 12 H, 7″,9″-H), 1.81 (s, 12 H, 8′,9′-H); δC (100 MHz, CD2Cl2/CD3OD) 161.1, 150.3, 149.9, 146.2, 145.1, 144.5, 139.8, 139.7, 139.6, 139.1, 138.3, 137.9, 137.8, 136.7, 136.1, 135.0, 134.2, 134.0, 132.1, 130.9, 129.8, 129.4, 128.6, 128.2, 127.9, 127.6, 127.5, 126.1, 125.4, 121.8, 120.6, 120.0, 119.4, 119.3, 105.7, 96.1, 87.7, 21.7, 21.4, 21.1, 21.1, 20.4, 18.6; ESI-MS m/z (%):1825.8 (75) [M + H]+, 913.9 (100) [M + 2H]2+; Found: C, 76.25; H, 5.31; N, 5.93. Calcd for C116H94Br2N8Zn: C, 76.33; H, 5.19; N, 6.14%
Using porphyrin P4 (105 mg, 88 µmol), Phen1 (200 mg, 375 µmol), Pd2(dba)3 (21 mg, 2.2 µmol), AsPh3 (30 mg, 98 µmol). Yield of 4: 130 mg (51%, 46 µmol); δH (400 MHz, CD2Cl2) 8.93 (s, 8 H, 2,3,7,8,12,13,17,18-Hβ), 8.63 (s, 4 H, 4-H), 8.32 (d, J = 8 Hz, 4 H, 7-H), 8.13 (d, J = 8 Hz, 8 H, Ar-Hb,Hb′), 7.89 (br s, 8 H, 5,6-H), 7.58 (d, J = 8 Hz, 4 H, 8-H), 7.53 (d, J = 8 Hz, 8 H, Ar-Ha,Ha′), 6.98 (s, 8 H, 3‴,5‴-H), 2.55 (s, 24 H, 8″,9″-H), 2.35 (s, 12 H, 8‴-H), 2.14 (s, 24 H, 7‴,9‴-H), 2.07 (s, 24 H, 7″,10″-H).; δC (100 MHz, CD2Cl2) 162.7, 160.9, 150.3, 146.4, 145.3, 143.7, 139.8, 139.0, 138.4, 137.9, 136.4, 136.1, 134.9, 134.3, 133.9, 132.3, 130.1, 129.3, 129.2, 128.6, 128.5, 128.0, 127.5, 127.4, 126.0, 125.5, 125.2, 122.3, 120.7, 120.2, 95.7, 87.9, 21.2, 21.1, 20.4, 18.6; ESI-MS m/z (%): 1402.9 (100) [M + 2H]2+, 936.1 (75) [M + 3H]3+, 702.0 (25) [M + 4H]4+; Found: C, 74.25; H, 5.08; N, 5.80. Calcd. for C176H136Br4N12Zn·2H2O: C, 74.43; H, 4.97; N, 5.92%
Triad 6. δH (400 MHz, CD2Cl2) 8.99 (s, 1 H, 4-H), 8.76 (d, J = 8 Hz, 1 H, 7-H), 8.74 (d, J = 4 Hz, 2 H, 2,18-Hβ), 8.69 (d, J = 4 Hz, 2 H, 3,17-Hβ), 8.66 (br s, 4 H, 7,8,12,13-Hβ), 8.53 (d, 4J = 2 Hz, 2 H, 2′,9′-H), 8.45 (d, 4J = 2 Hz, 2 H, 4′,7′-H), 8.29 (d, J = 9 Hz, 1 H, 5-H ), 8.26 (d, J = 9 Hz, 1 H, 6-H), 8.13 (d, J = 8 Hz, 2 H, Ar-Ha,Ha′), 7.97 (d, J = 8 Hz, 1 H, 8-H), 7.88 (s, 2 H, 5′,6′-H), 7.48 (d, J = 8 Hz, 2 H, Ar-Hb,Hb′), 7.23 (s, 6 H, 3⁗,5⁗-H), 6.16 (s, 2 H, 3‴,5‴-H), 4.57 (d, J = 5 Hz, 4 H, 2,5,2″,5″-Hcp), 4.35 (d, J = 3 Hz, 4 H, 3,4,3″,4″-Hcp), 4.26 (s, 10 H, 1′,2′,3′,4′,5′,1‴,2‴,3‴,4‴,5‴-Hcp) 2.58 (s, 6 H, por-mes-Me ), 2.56 (s, 3H, por-mes-Me), 1.95 (s, 6 H, 8″,9″-H), 1.93 (s, 6 H, 7″,10″), 1.79 (s, 12 H, por-mes-Me), 1.77 (s, 6 H, 7‴,9‴-H), 1.76 (s, 6 H, por-mes-Me),1.64 (s, 3 H, 8‴-H); δC (100 MHz, CD2Cl2) 161.3, 159.9, 150.3, 150.1, 149.9, 149.8, 144.1, 142.6, 140.8, 139.7, 139.6, 139.5, 139.4, 138.3, 138.0, 137.8, 137.7, 137.3, 135.2, 135.0, 133.9, 132.8, 131.8, 131.4, 131.3, 130.9, 130.0, 129.3, 128.6, 128.5, 128.2, 128.1, 127.9, 127.6 127.4, 127.3, 126.7, 123.3, 122.5, 120.9, 119.0, 118.7, 116.9, 98.0, 96.9, 86.1, 81.8, 72.3, 70.5, 70.3, 21.8, 21.5, 20.6, 20.4, 20.3, 18.8; ESI-MS m/z (%): 1995.7(100) [M]+. Found: C, 65.88; H, 4.36; N, 5.65. Calcd for C122H97BrCuF6Fe2N8PZn·CH2Cl2: C, 66.38; H, 4.48; N, 5.03%.
Triad 7. δH (400 MHz, CD2Cl2) 9.00 (s, 2 H, 4-H), 8.77 (d, J = 8 Hz, 2 H, 7-H), 8.76 (s, 2 H, 2,3-Hβ), 8.73 (d, J = 4 Hz, 2 H, 7,18-Hβ), 8.69 (d, J = 4 Hz, 2 H, 8,17-Hβ), 8.68 (s, 2 H, 12,13-Hβ), 8.53 (d, J = 2 Hz, 4 H, 2′,9′-H), 8.46 (s, 4 H, 4′,7′-H), 8.31 (d, J = 8 Hz, 2 H, 5-H), 8.28 (d, J = 8 Hz, 2 H, 6-H), 8.06 (d, J = 8 Hz, 4 H, Ar-Ha,Ha′), 7.97 (d, J = 8 Hz, 2 H, 8-H), 7.89 (s, 4 H, 5′,6′-H), 7.48 (d, J = 8 Hz, 4 H, Ar-Hb,Hb′), 7.26 (s, 4 H, 3⁗,5⁗-H), 6.16 (s, 4 H, 3‴,5‴-H), 4.60 (s, 8 H, 2,5,2″,5″-Hcp), 4.38 (s, 8 H, 3,4,3″,4″-Hcp), 4.29 (s, 20 H, 1′,2′,3′,4′,5′,1‴,2‴,3‴,4‴,5‴-Hcp), 2.58 (s, 6 H, por-mes-Me), 1.94 (s, 12 H, 8″,9″-H), 1.93 (s, 12 H, 7″,9″-H), 1.79 (s, 12 H, 7‴,9‴-H ), 1.77 (s, 12 H, por-mes-Me), 1.64 (s, 6 H, 8‴-H); δC (100 MHz, CD2Cl2), 161.2, 159.8, 150.6, 150.2, 149.9, 149.7, 144.5, 144.1, 142.6, 140.8, 139.7, 139.4, 139.0, 138.3, 138.1, 137.9, 137.7, 137.3, 135.2, 134.9, 133.9, 132.8, 131.7, 131.3, 130.1, 129.3, 128.7, 128.6, 128.5, 128.2, 128.0, 127.9, 127.6, 127.4, 127.3, 126.7, 123.2, 122.5, 121.2, 119.9, 119.5, 116.9, 97.8, 96.9, 86.2, 81.8, 77.9, 72.3, 70.6, 70.4, 63.4, 21.7, 21.5, 20.6, 20.4, 20.3, 18.8; ESI-MS m/z (%): 1572.3 (100) [M]2+ Found: C, 60.68; H, 3.93; N, 4.54 Calcd. for C188H142Br2Cu2F12Fe4N12P2Zn·4CH2Cl2: C, 61.09; H, 4.01; N, 4.45%
Triad 8. δH (400 MHz, CD2Cl2) 8.98 (s, 2 H, 4-H), 8.77 (d, J = 8 Hz, 2 H, 7-H), 8.75 (d, J = 4 Hz, 4 H, 2,8,12,18-Hβ), 8.69 (d, J = 4 Hz, 4 H, 3,7,13,17-Hβ), 8.49 (s, 4 H, 2′,9′-H), 8.41 (s, 4 H, 4′,7′-H), 8.29 (d, J = 8 Hz, 2 H, 5-H ), 8.28 (d, J = 8 Hz, 2 H, 6-H), 8.08 (d, J = 8 Hz, 4 H, Ar-Ha,Ha′), 7.96 (d, J = 8 Hz, 2 H, 8-H), 7.86 (s, 4 H, 5′,6′-H), 7.48 (d, J = 7.8 Hz, 4 H, Ar-Hb,Hb′), 7.25 (s, 4 H, 3⁗,5⁗-H), 6.14 (s, 4 H, 3‴,5‴-H), 4.70 (br s, 8 H, 2,5,2″,5″-Hcp), 4.43 (br s, 28 H, 3,4,3″,4″-1′,2′,3′,4′,5′,1‴,2‴,3‴,4‴,5‴-Hcp), 2.59 (s, 6 H, por-mes-Me), 1.92 (s, 12 H, 8″,9″-H), 1.90 (s, 12 H, 7″,10″-H), 1.77 (s, 12 H, 7‴,9‴-H), 1.75 (s, 12 H, por-mes-Me), 1.62 (s, 6 H, 8‴-H); δC (100 MHz, CD2Cl2) 161.3, 159.8, 149.9, 144.1, 142.6, 140.7, 139.6, 139.4, 139.0, 138.3, 138.0, 137.9, 137.7, 137.3, 135.1, 134.9, 133.9, 132.7, 132.3, 131.2, 130.2, 130.1, 129.3, 128.8, 128.6, 128.5, 128.2, 128.0, 127.9, 127.7, 127.6, 127.4, 126.6, 123.2, 122.5, 121.2, 116.8, 97.0, 86.2, 81.7, 73.1, 71.8, 30.0, 21.6, 21.5, 20.6, 20.4, 20.3, 18.8; ESI-MS m/z (%): 1572.3 (100) [M2+]; Found: C, 61.04; H, 4.08; N, 4.42; Calcd. for: C188H142Br2Cu2F12Fe4N12P2Zn·4CH2Cl2: C, 61.09; H, 4.01; N, 4.45%.
Triad 9. δH (400 MHz, CD2Cl2) 8.99 (s, 4 H, 4-H), 8.78 (d, J = 8 Hz, 4 H, 7-H), 8.75 (s, 8 H, Hβ), 8.51 (s, 8 H, 2′,9′-H), 8.44 (s, 8 H, 4′,7′-H), 8.30 (br s, 8 H, 5,6-H), 8.02 (d, J = 8 Hz, 8 H, Ar-Ha,Ha′), 7.98 (d, J = 8.0 Hz, 4 H, 8-H), 7.88 (s, 8 H, 5′,6′-H), 7.46 (d, J = 8 Hz, 8 H, Ar-Hb,Hb′), 6.14 (s, 8 H, 3‴,5‴-H), 4.64 (s, 16 H, 2,5,2″,5″-Hcp), 4.42 (s, 16 H, 3,4,3″,4″-Hcp), 4.32 (s, 40 H, 1′,2′,3′,4′,5′,1‴,2‴,3‴,4‴,5‴-Hcp), 1.92 (s, 48 H, 7″,8″,9″,10″-H), 1.76 (s, 24 H, 7‴,9‴-H), 1.63 (s, 12 H, 8‴-H); δC (100 MHz, CD2Cl2) 161.1, 159.7, 150.1, 149.9, 144.1, 142.6, 140.7, 139.8, 139.4, 138.3, 137.9, 137.6, 137.4, 135.1, 134.8, 133.8, 132.7, 132.2, 130.1, 129.2, 128.6, 128.5, 128.1, 128.0, 127.6, 127.4, 126.7, 126.7, 123.1, 122.4, 116.8, 97.5, 96.9, 86.2, 81.8, 72.8, 71.1, 30.0, 20.6, 20.4, 20.3, 18.8; ESI-MS m/z (%): [M]4+ 1360.5 (100); Found: C, 60.76; H, 4.16; N, 4.47; Calcd. for C320H232Br4Cu4F24Fe8N20P4Zn·4CH2Cl2: C, 61.16; H, 3.80; N, 4.40%
Tetrad 11. δH (400 MHz, CD2Cl2–CS2) 8.97 (s, 1 H, 4-H), 8.81 (d, J = 8 Hz, 1 H, 7-H), 8.63 (d, J = 4 Hz, 2 H, 2,18-Hβ), 8.57 (d, J = 4 Hz, 2 H, 3,17-Hβ), 8.55 (br s, 4 H, 7,8,12,13-Hβ), 8.54 (d, 4J = 2 Hz, 2 H, 2′,9′-H), 8.44 (d, 4J = 2 Hz, 2 H, 4′,7′-H), 8.35 (d, J = 9 Hz, 1 H, 5-H ), 8.30 (d, J = 9 Hz, 1 H, 6-H), 8.04 (d, J = 8 Hz, 2 H, Ar-Ha,Ha′), 7.96 (d, J = 8 Hz, 1 H, 8-H), 7.89 (s, 2 H, 5′,6′-H), 7.41 (d, J = 8 Hz, 2 H, Ar-Hb,Hb′), 7.20 (s, 6 H, 3⁗,5⁗-H), 6.45 (br s, 2 H, 3,5-Hpy), 6.20 (s, 2 H, 3‴,5‴-H), 4.66 (d, J = 9 Hz, 1 H, pyr-H5a), 4.58 (s, 4 H, 2,5,2″,5″-Hcp), 4.56 (s, 2 H, pyr-H2), 4.36 (s, 4 H, 3,4,3″,4″-Hcp), 4.27 (s, 10 H, 1′,2′,3′,4′,5′,1‴,2‴,3‴,4‴,5‴-Hcp), 3.92 (d, J = 9 Hz, 1 H, pyr-H5b), 2.59 (s, 9 H, por-mes-Me), 1.97 (s, 6 H, 8″,9″-H), 1.93 (s, 6 H, 7″,10″-H),1.82 (s, 6 H, 7‴,9‴-H), 1.73 (s, 18 H, por-mes-Me), 1.71 (s, 3 H, 8‴-H), 1.27 (s, 3 H, N–CH3–C60); δC (100 MHz, CD2Cl2) 206.1, 192.8, 161.0, 159.6, 155.8, 153.5, 151.8, 151.2, 149.9, 149.8, 149.6, 149.3, 147.4, 146.3 (2), 146.2, 146.0, 145.7, 145.5(2), 145.4, 145.3, 145.2, 145.1, 144.7, 144.5, 144.4, 144.3, 144.0, 143.1, 143.0, 142.7, 142.6, 142.5, 142.2 (2), 142.1, 142.0, 141.9, 141.8, 141.7, 141.6, 141.5, 140.6, 140.2, 139.7(2), 139.6, 139.4, 139.2, 138.1, 137.8, 137.6, 137.3, 137.2, 136.7, 136.1, 135.9, 135.5, 135.0, 133.9, 132.7, 131.6, 131.2, 131.0, 130.6, 129.8, 129.2, 128.5, 128.4, 128.0, 127.9, 127.8, 127.5, 127.4, 127.3, 127.2, 126.6, 123.1, 122.4, 120.5, 118.7, 118.6, 118.3, 97.9, 96.7, 81.8, 80.9, 72.3, 70.5, 70.3, 54.3, 39.1, 30.8, 21.9, 21.8, 21.4, 20.3, 20.2, 18.7. Found: C, 73.85; H, 3.75; N, 4.41. Calcd. for C190H107BrCuF6Fe2N10PZn·1½CH2Cl2: C, 73.65; H, 3.55; N, 4.49%.
Tetrad 12. δH (400 MHz, C2D4Cl2) 8.98 (s, 2 H, 4-H), 8.75 (d, J = 8 Hz, 2 H, 7-H), 8.62 (s, 2 H, 2,3-Hβ), 8.59 (d, J = 4 Hz, 2 H, 7,18-Hβ), 8.54 (d, J = 4 Hz, 2 H, 8,17-Hβ), 8.51 (s, 2 H, 12,13-Hβ), 8.47 (d, J = 2 Hz, 4 H, 2′,9′-H), 8.44 (d, J = 2 Hz, 4 H, 4′,7′-H), 8.29 (d, J = 8 Hz, 2 H, 5-H), 8.28 (d, J = 8 Hz, 2 H, 6-H), 7.97 (d, J = 8 Hz, 4 H, Ar-Ha,-Ha′), 7.95 (d, J = 8 Hz, 2 H, 8-H), 7.85 (s, 4 H, 5′,6′-H), 7.42 (d, J = 8 Hz, 4 H, Ar-Hb,Hb′), 7.16 (s, 4 H, 3⁗,5⁗-H), 6.26 (br s, 2 H, 3,5-Hpy), 6.07 (s, 4 H, 3‴,5‴-H), 4.57 (s, 8 H, 2,5,2″,5″-Hcp), 4.36 (s, 1 H, pyr-H2), 4.33 (s, 8 H, 3,4,3″,4″-Hcp), 4.24 (s, 20 H, 1′,2′,3′,4′,5′,1‴,2‴,3‴,4‴,5‴-Hcp), 4.22 (d, J = 9 Hz, 1 H, pyr-H5b), 2.52 (s, 6 H, por-mes-Me), 1.91 (s, 12 H, 8″,9″-H), 1.87 (s, 12 H, 7″,10″-H), 1.72 (s, 12 H, 7‴,9‴-H), 1.64 (s, 12 H, por-mes-Me), 1.56 (s, 6 H, 8‴-H), 1.23 (s, 3 H, N–CH3–C60). δC (100 MHz, C2D4Cl2) 206.0, 192.7, 160.9, 159.5, 155.7, 153.4, 151.7, 151.2, 150.1, 149.9, 149.7, 149.6, 149.3, 146.2, 146.0, 145.4(2), 145.3(2), 145.2, 145.1, 145.0, 144.9, 144.6, 144.0, 142.8, 142.6, 142.5, 142.4, 142.0, 141.9, 141.8, 141.7, 141.5, 140.6, 140.1, 139.7, 139.4, 139.2, 138.1, 137.8, 137.6, 137.5, 137.2, 135.0, 133.8, 132.9, 131.8, 131.3, 130.9, 129.8, 129.1, 128.5, 128.4, 128.0, 127.9, 127.8, 127.5, 127.3, 126.6, 123.0, 122.4, 120.6, 119.3, 118.9, 97.7, 96.7, 86.2, 81.8, 72.3, 70.6, 70.3, 63.4, 54.3, 39.1, 30.8, 21.9, 21.4, 20.4, 20.3, 20.2, 18.7 ESI-MS m/z (%): [M]2+ 1999.0 (20); Found: C, 65.61; H, 3.53; N, 3.97; Calcd. for C256H152Br2Cu2F12Fe4N14P2Zn·6CH2Cl2: C, 65.57; H, 3.44; N, 4.09%.
Tetrad 13. δH (400 MHz, C2D4Cl2) 8.99 (s, 2 H, 4-H), 8.77 (d, J = 8 Hz, 2 H, 7-H), 8.63 (d, J = 4 Hz, 4 H, 2,8,12,18-Hβ), 8.56 (d, J = 4 Hz, 4 H, 3,7,13,17-Hβ), 8.48 (s, 4 H, 2′,9′-H), 8.45 (s, 4 H, 4′,7′-H), 8.29 (d, J = 8 Hz, 2 H, 5-H), 8.28 (d, J = 8 Hz, 2 H, 6-H), 7.98 (d, J = 8 Hz, 4 H, Ar-Ha,Ha′), 7.97 (d, J = 8 Hz, 2 H, 8-H), 7.87 (s, 4 H, 5′,6′-H), 7.44 (d, J = 7.8 Hz, 4 H, Ar-Hb,Hb′), 7.19 (s, 6 H, 3⁗,5⁗-H), 6.22 (br s, 2 H, 3,5-Hpy), 6.08 (s, 4 H, 3‴,5‴-H), 4.63 (s, 8 H, 2,5,2″,5″-Hcp), 4.57 (d, J = 9 Hz, 1 H, pyr-H5a), 4.45 (s, 1 H, pyr-H2), 4.38 (s, 8 H, 3,4,3″,4″-Hcp), 4.29 (s, 20 H, 1′,2′,3′,4′,5′,1‴,2‴,3‴,4‴,5‴-Hcp), 3.83 (d, J = 9 Hz, 1 H, pyr-H5b), 2.54 (s, 6 H, por-mes-Me), 1.94 (s, 12 H, 8″,9″-H), 1.89 (s, 12 H, 7″,10″-H), 1.75 (s, 12 H, 7‴,9‴-H), 1.67 (s, 12 H, por-mes-Me), 1.57 (s, 6 H, 8‴-H), 1.25 (s, 3 H, N–CH3–C60); δC (100 MHz, C2D4Cl2) 206.1, 192.8, 160.9, 159.6, 155.8, 153.5, 149.9, 146.3, 146.2, 146.0, 145.5(2), 145.4, 145.2, 144.8, 144.5, 144.4, 144.3, 144.0, 142.8, 142.7, 142.6, 142.5, 142.2(2), 142.1, 141.8, 141.5, 140.6, 140.2, 139.7, 139.4, 139.3, 139.2, 138.1, 137.8, 137.6, 137.5, 137.2, 136.2, 135.9, 135.5, 135.0, 133.9, 132.8, 132.0, 130.8, 129.9, 129.2, 128.5, 128.4, 128.0, 127.9, 127.8, 127.5, 127.3, 127.2, 126.5, 123.0, 122.4, 120.6, 97.8, 96.7, 96.2, 81.8, 72.5, 70.8, 70.6, 54.3, 39.1, 30.8, 21.8, 21.4, 20.3, 20.2, 18.7. ESI-MS m/z (%): [M]2+ 1999.5 (60); Found: C, 67.49; H, 3.65; N, 4.15. Calcd. for C256H152Br2Cu2F12Fe4N14P2Zn)·4CH2Cl2: C, 67.46; H, 3.48; N, 4.24%.
Tetrad 14. δH (400 MHz, CD2Cl2–CS2): 8.99 (s, 4 H, 4-H), 8.78 (d, J = 8 Hz, 4 H, 7-H), 8.67 (s, 8 H, Hβ), 8.51 (s, 8 H, 2′,9′-H), 8.44 (s, 8 H, 4′,7′-H), 8.32 (s, 8 H, 5,6-H), 7.96 (d, J = 8 Hz, 8 H, Ar-Ha,Ha′), 7.94 (d, J = 8.0 Hz, 4 H, 8-H), 7.89 (s, 8 H, 5′,6′-H), 7.40 (d, J = 8 Hz, 8 H, Ar-Hb,Hb′), 6.23 (br s, 2 H, 3,5-Hpy), 6.18 (s, 8 H, 3‴-H, 5‴-H), 4.77 (d, J = 9 Hz, 1 H, pyr-H5a), 4.62 (s, 16 H, 2,5,2″,5″-Hcp), 4.46 (s, 1 H, pyr-H2), 4.40 (s, 16 H, 3,4,3″,4″-Hcp), 4.30 (s, 40 H, 1′,2′,3′,4′,5′,1‴,2‴,3‴,4‴,5‴-Hcp), 3.95 (d, J = 9 Hz, 1 H, pyr-H5b),1.94 (s, 24 H, 7″,8″-H) 1.90 (s, 24 H, 9″,10″-H), 1.78 (s, 24 H, 7‴,9‴-H), 1.68 (s, 12 H, 8‴-H), 1.28 (s, 3H, N-CH3); δC (100 MHz, C2D4Cl2) 206.0, 192.8, 149.9, 147.4, 146.2, 146.1, 145.9, 145.5, 145.3, 144.3, 142.5, 141.9, 140.6, 140.1, 139.8, 139.5, 138.1, 137.6, 137.2, 136.4, 135.9, 135.0, 134.9, 134.9, 134.1, 133.8, 132.8, 131.8, 129.8, 128.6, 128.4, 128.0, 127.9, 127.5, 127.3, 127.2, 122.4, 120.8, 96.7, 86.2, 81.8, 72.3, 70.5, 70.4, 70.3, 63.3, 54.3, 39.1, 30.8, 21.1, 20.4, 20.3, 20.2, 18.7; Found: C, 61.89; H, 3.59; N, 3.91 Calcd. for C388H242Br4Cu4F24Fe8N22P4Zn·10CH2Cl2 : C, C, 61.86; H, 3.42; N, 3.99%.
Acknowledgements
This publication is dedicated to Professor Dr Manfred Christl (Universität Würzburg) on the occasion of his 65th birthday. We are grateful to the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie for continued financial support.References
- The Photosynthetic Reaction Center, ed. J. Deisenhofer and J. R. Norris, Academic Press, New York, 1993 Search PubMed.
- Electron Transfer in Chemistry Vol. I–V, ed. V. Balzani, Wiley-VCH, Weinheim, 2001 Search PubMed.
- (a) J. R. Winkler and H. B. Gray, Chem. Rev., 1992, 92, 369 CrossRef; (b) C. C. Moser, J. M. Keske, K. Warncke, R. S. Farid and P. L. Dutton, Nature, 1992, 355, 796 CrossRef CAS; (c) C. Kirmaier and D. Holton, in The Photosynthetic Reaction Center, ed. J. Deisenhofer and J. R. Norris, Academic Press, San Diego, 1993, vol. II, p. 49 Search PubMed; (d) R. Langen, I.-J. Chang, J. P. Germanas, J. H. Richards, J. R. Winkler and H. B. Gray, Science, 1995, 268, 1733 CAS; (e) C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47 CrossRef CAS.
- (a) D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695 CrossRef CAS; (b) D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22 RSC; (c) M. D. Meijer, G. P. M. van Klink and G. van Koten, Coord. Chem. Rev., 2002, 230, 141 CrossRef; (d) M. E. El-Khouly, O. Ito, P. M. Smith and F. D'Souza, J. Photochem. Photobiol., C, 2004, 5, 79 CrossRef CAS; (e) Molecular Devices and Machines, ed. V. Balzani, M. Venturi and A. Credi, Wiley-VCH, Weinheim, 2003 Search PubMed; (f) Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001 Search PubMed; (g) Electron Transfer, ed. J. Jortner and M. Bixon, John Wiley & Sons, New York, 1999, parts 1 and 2 Search PubMed; (h) Molecular Electronics, ed. J. Jortner and M. Ratner, Blackwell Science, Oxford, 1997 Search PubMed; (i) Organic Photovoltaics, ed. C. Brabec, V. Dyakonov, J. Parisi and N. S. Sariciftci, Springer, Berlin, 2003 Search PubMed; (j) K. Domen, J. N. Kondo, H. Hara and T. Takata, Bull. Chem. Soc. Jpn., 2000, 73, 1307 CrossRef CAS.
- (a) H. Imahori, Org. Biomol. Chem., 2004, 2, 1425 RSC; (b) P. A. Liddell, D. Kuciauskas, J. P. Sumida, B. Nash, D. Nguyen, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 1997, 119, 1400 CrossRef CAS; (c) P. A. Liddell, G. Kodis, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 2002, 124, 7668 CrossRef CAS; (d) F. D'Souza, G. R. Deviprasad, M. E. Zandler, M. E. El-Khouly, M. Fujitsuka and O. Ito, J. Phys. Chem. B, 2002, 106, 4952 CrossRef CAS; (e) F. D'Souza, P. M. Smith, M. E. Zandler, A. L. McCarty, M. Itou, Y. Araki and O. Ito, J. Am. Chem. Soc., 2004, 126, 7898 CrossRef CAS; (f) K. Li, D. I. Schuster, D. M. Guldi, M. A. Herranz and L. Echegoyen, J. Am. Chem. Soc., 2004, 126, 3388 CrossRef CAS; (g) S. N. Smirnov, P. A. Liddell, I. V. Vlassiouk, A. Teslja, D. Kuciauskas, C. L. Braun, A. L. Moore, T. A. Moore and D. Gust, J. Phys. Chem. A, 2003, 107, 7567 CrossRef CAS.
- (a) S. Fukuzumi, H. Imahori, H. Yamada, M. E. El-Khouly, M. Fujitsuka, O. Ito and D. M. Guldi, J. Am. Chem. Soc., 2001, 123, 2571 CrossRef CAS; (b) H. Imahori, K. Tamaki, D. M. Guldi, C. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607 CrossRef CAS.
- H. Imahori, Y. Sekiguchi, Y. Kashiwagi, T. Sato, Y. Araki, O. Ito, H. Yamada and S. Fukuzumi, Chem. Eur. J., 2004, 10, 3184 CrossRef CAS.
- (a) T. Hayashi and H. Ogoshi, Chem. Soc. Rev., 1997, 26, 355 RSC; (b) M. W. Ward, Chem. Soc. Rev., 1997, 26, 365 RSC; (c) P. Piotrowiak, Chem. Soc. Rev., 1999, 28, 143 RSC.
- (a) F. D'Souza, P. M. Smith, S. Gadde, A. L. McCarty, M. J. Kullman, M. E. Zandler, M. Itou, Y. Araki and O. Ito, J. Phys. Chem. B, 2004, 108, 11333 CrossRef CAS; (b) H. Nakagawa, K. Ogawa, A. Satake and Y. Kobuke, Chem. Commun., 2006, 1560 RSC.
- (a) J.-C. Chambron, J.-P. Collin, J.-O. Dalbavie, C. O. Dietrich-Buchecker, V. Heitz, F. Odobel, N. Solladié and J.-P. Sauvage, Coord. Chem. Rev., 1998, 1299 CrossRef CAS; (b) M. Andersson, M. Linke, J.-C. Chambron, J. Davidsson, V. Heitz, L. Hammarström and J.-P. Sauvage, J. Am. Chem. Soc., 2002, 124, 4347 CrossRef CAS.
- N. Armaroli, Chem. Soc. Rev., 2001, 30, 113 RSC.
- M. Schmittel, C. Michel, A. Wiegrefe and V. Kalsani, Synthesis, 2001, 1561 CrossRef CAS.
- (a) M. Schmittel and R. S. K. Kishore, Org. Lett., 2004, 6, 1923 CrossRef CAS; (b) R. S. K. Kishore, T. Paululat and M. Schmittel, Chem. Eur. J., 2006, 12, 8136 CrossRef; (c) R. S. K. Kishore, K. Venkateshwaralu and M. Schmittel, Chem. Commun., 2006, 3690 RSC.
- (a) J. S. Lindsey, S. Prathapan, T. E. Johnson and R. W. Wagner, Tetrahedron, 1994, 50, 8941 CrossRef CAS; (b) J. K. Laha, S. Dhanalekshmi, M. Taniguchi, A. Ambroise and J. S. Lindsey, Org. Process. Res. Dev., 2003, 7, 799 Search PubMed; (c) B. J. Littler, Y. Ciringh and J. S. Lindsey, J. Org. Chem., 1999, 64, 2864 CrossRef CAS; (d) W. Wagner, T. E. Johnson and J. S. Lindsey, J. Am. Chem. Soc., 1996, 118, 11166 CrossRef CAS.
- (a) HETPHEN: a quantitative approach to HETeroleptic bisPHENanthroline metal complexes. This approach utilises steric and electronic effects originating from bulky aryl substituents at the bis-imine coordination sites to control the coordination equilibrium both kinetically and thermodynamically. M. Schmittel and A. Ganz, Chem. Commun., 1997, 999 Search PubMed; (b) M. Schmittel, U. Lüning, M. Meder, A. Ganz, C. Michel and M. Herderich, Heterocycl. Commun., 1997, 3, 493 CAS.
- F. D'Souza, G. R. Deviprasad, M. E. Zandler, V. T. Hoang, K. Arkady, M. Van Stipdonk, A. Perera, M. E. El-Khouly, M. Fujitsuka and O. Ito, J. Phys. Chem. A, 2002, 106, 3243 CrossRef CAS.
- (a) M. Nappa and J. S. Valentine, J. Am. Chem. Soc., 1978, 100, 5075 CrossRef CAS; (b) F. D'Souza, Y.-Y. Hsieh and G. R. Deviprasad, Inorg. Chem., 1996, 35, 5747 CrossRef CAS.
- (a) H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 95 CrossRef CAS; (b) J. C. Rossoti, H. S. Rossoti and R. J. Whewell, J. Inorg. Nucl. Chem., 1971, 33, 2051 CrossRef CAS; (c) H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 257 CAS; (d) H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1986, 33, 943 CrossRef CAS.
- SPARTAN, Wavefunction, Inc., 18401 Von Karman Avenue, Suite 370, Irvine, CA 92612, USA Search PubMed.
- K. Funatsu, T. Imamura, A. Ichimura and Y. Sasaki, Inorg. Chem., 1998, 37, 1798 CrossRef CAS.
- (a) L. Flamigni, A. M. Talarico, J.-C. Chambron, V. Heitz, M. Linke, N. Fujita and J.-P. Sauvage, Chem. Eur. J., 2004, 10, 2689 CrossRef CAS; (b) J.-C. Chambron, A. Harriman, V. Heitz and J.-P. Sauvage, J. Am. Chem. Soc., 1993, 115, 6109 CrossRef.
- G. M. Sheldrick, SADABS, University of Göttingen, Germany, 2000 Search PubMed.
- G. M. Sheldrick, Programs for the Solution and Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S27, Tables S1–S3, and crystal structure data for 5. See DOI: 10.1039/b614111k |
‡ Crystal structure: a single crystal (a brown blade with dimensions 0.04 × 0.26 × 0.75 mm) was measured on a SIEMENS SMART 1 K CCD diffractometer with Mo-Kα radiation at a temperature of about 158 K. Repeatedly measured reflections remained stable. An empirical absorption correction with program SADABS22 gave a correction factor between 0.804 and 0.952. 49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 615 reflections measured, 9002 unique reflections, R(I)int = 0.063. The structure was determined by direct methods and refined by least-squares against all measured F2 values.23 The H atoms were positioned geometrically and were constrained. C36H24Fe2N2·CHCl3, M = 715.64, monoclinic, space group P21/c (no. 14), a = 22.770(3), b = 7.5283(11), c = 19.570(3) Å, β = 111.923(12)°, V = 3112.1(8) Å3, Z = 4, Dx = 1.527 g cm−3, µ = 1.22 mm−1, 397 refined parameters, R1(F)[I > 2σ(I)] = 0.052, GOOF = 1.02, the final difference density was between −1.12 and +1.07 e Å−3 near Cl atoms of the chloroform solvate group. CCDC reference number 622456. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b614111k 615 reflections measured, 9002 unique reflections, R(I)int = 0.063. The structure was determined by direct methods and refined by least-squares against all measured F2 values.23 The H atoms were positioned geometrically and were constrained. C36H24Fe2N2·CHCl3, M = 715.64, monoclinic, space group P21/c (no. 14), a = 22.770(3), b = 7.5283(11), c = 19.570(3) Å, β = 111.923(12)°, V = 3112.1(8) Å3, Z = 4, Dx = 1.527 g cm−3, µ = 1.22 mm−1, 397 refined parameters, R1(F)[I > 2σ(I)] = 0.052, GOOF = 1.02, the final difference density was between −1.12 and +1.07 e Å−3 near Cl atoms of the chloroform solvate group. CCDC reference number 622456. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b614111k |
| This journal is © The Royal Society of Chemistry 2007 |