Marine natural products: synthetic aspects
Jonathan C.
Morris
a,
Gillian M.
Nicholas
b and
Andrew J.
Phillips
*c
aSchool of Chemistry and Physics, University of Adelaide, Adelaide, Australia 5005
bRoche Colorado Corporation, 2075 North 55th St., Boulder, Colorado 80301, USA
cDepartment of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309, USA. E-mail: Andrew.Phillips@colorado.edu; Fax: +1 303 492 0439; Tel: +1 303 735 2049
First published on 4th January 2007
Abstract
Covering: January to December 2005. Previous review: Nat. Prod. Rep., 2006, 23, 79
An overview of marine natural products synthesis during 2005 is provided. In a similar vein to earlier installments in this series, the emphasis is on total syntheses of molecules of contemporary interest, new total syntheses, and syntheses that have resulted in structure confirmation or stereochemical assignments.
 Jonathan Morris | Jonathan Morris obtained his B.Sc. (Hons) degree from the University of Western Australia and his Ph.D. degree from The Australian National University in Canberra, Australia. After a postdoctoral appointment with Phil Magnus at the University of Texas at Austin, he took up his first academic position at the University of Canterbury, New Zealand. In 2004, he moved to the University of Adelaide. His research interests focus around the synthesis of biologically active natural products. |
 Gillian Nicholas | Gillian Nicholas received her B.Sc., M.Sc., and Ph.D. degrees from the University of Canterbury, where she worked on the isolation and structure elucidation of biologically active compounds from fungal sources with John Blunt and Murray Munro. Postdoctoral research with Ted Molinski at UC Davis and Carole Bewley at the National Institutes of Health was followed by a 2 year stay at Hauser Inc. in Denver, CO. She is currently a Principal Analytical Research Chemist at Roche Colorado Corporation in Boulder, CO. |
 Andy Phillips | Andy Phillips obtained his B.Sc. (Hons) and Ph.D. degrees from the University of Canterbury in Christchurch, New Zealand. After a postdoctoral appointment with Peter Wipf at the University of Pittsburgh, he joined the faculty at the University of Colorado. His research interests are broadly defined by the development of new methods and strategies for the synthesis of complex natural products. |
1 Introduction
This review is designed to provide an overview of key features of the 2005 literature covering the synthesis of marine natural products and should act as a companion to the Marine Natural Products review published in this journal.1 The emphasis is on total syntheses of molecules of contemporary interest. Tabulated data for other syntheses are also provided.2 Review articles
A number of reviews that cover various aspects of marine natural products synthesis have appeared: “Advances in the total synthesis of biologically important marine macrolides”,2 “Asymmetric total synthesis of complex marine natural products”,3 “Challenge palau'amine: current standings”,4 “Convergent strategies for syntheses of trans-fused polycyclic ethers”,5 “Development of new synthetic methods and its application to total synthesis of nitrogen-containing bioactive natural products”,6 “Discovery of E7389, a fully synthetic macrocyclic ketone analog of halichondrin B”,7 “Ecteinascidin 743 (ET-743; Yondelis), aplidin, and kahalide F”,8 “Joys of molecules. 1. Campaigns in total synthesis”,9 “Large-scale production of pharmaceuticals by marine sponges: sea, cell, or synthesis?”,10 “Marine natural products”,11 “Marine natural products as anticancer drugs”,12 “Marine natural products from Pseudopterogorgia elisabethae: structures, biosynthesis, pharmacology, and total synthesis”,13 “Novel antitumor agents: marine sponge alkaloids, their synthetic analogs and derivatives”,14 “Progress in the total synthesis of laurencin and prelaureatin”,15 “Recent progress in the total synthesis of dolabellane and dolastane diterpenes”,16 “Recent progress of the synthetic studies of biologically active marine cyclic peptides and depsipeptides”,17 “The synthesis of aplysinopsins, meridianines, and related compounds”,18 “Synthetic chemistry of halichlorine and the pinnaic acids”,19 “Synthetic paths towards scalaranes: assembling the scalaranic skeleton and further transformations”,20 “Recent advances in natural product synthesis by using intramolecular Diels–Alder reactions”,21 and “Total synthesis of oxacyclic macrodiolide natural products”.22 Other reviews of relevance are cited in the text.3 Spongistatin-1
Ley and co-workers have reported a convergent total synthesis of spongistatin-1, 1.23 Recognising that the 42-membered macrolide had previously been assembled by a Wittig coupling and a C1–C41(OH) macrolactonization, the key intermediates were established to be ABCD fragment 2 and EF fragment 3 (Scheme 1).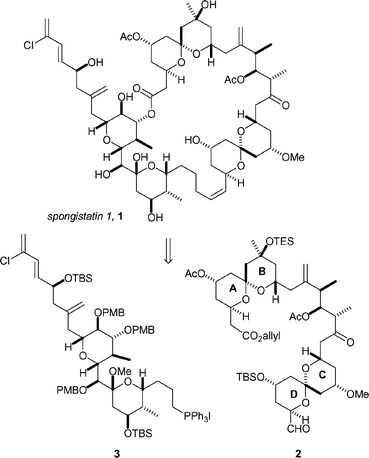 | ||
| Scheme 1 Ley's retrosynthetic analysis of spongistatin-1, 1. | ||
Although the Ley group had previously developed syntheses of the AB and CD fragments, the overall length of the synthesis was deemed to be too long.24 Recognizing that the ABCD fragment had a high level of latent C2-symmetry around C15, an efficient second-generation synthesis was developed (Scheme 2). The key to this was the realization that cyclization of an epimeric mixture of the acyclic precursor 4 would lead to the formation of the AB spiroketal core 5 and the CD spiroketal core 6.
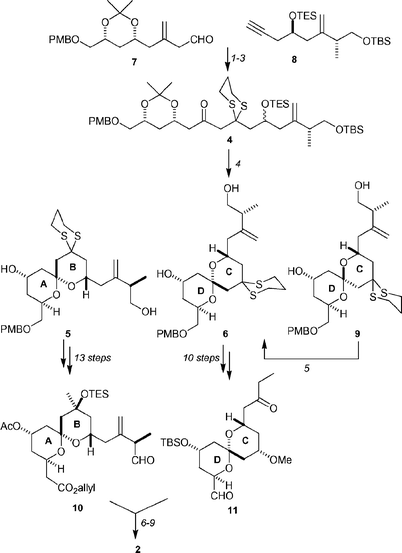 | ||
| Scheme 2 Synthesis of the ABCD fragment of spongistatin-1 (1). Reagents and conditions: (1) iPrMgCl, 8, THF, rt, then 7, THF, −20 °C; (2) Dess–Martin periodinane, CH2Cl2, rt, 85% over two steps; (3) HS(CH2)3SH, NaOMe, MeOH–CH2Cl2, −10 °C → rt, 97%; (4) 10% aq. HClO4, MeCN–CH2Cl2, rt, 86%, ratio 5 : 6 : 9 = 1.00 : 0.24 : 0.76; (5) 3.5% aq. HClO4, 5 equiv. Ca(ClO4)2·4H2O, MeCN–CH2Cl2, rt, 87% after three recycles; (6) Cy2BCl, NEt3, Et2O, −78 °C → 0 °C; 0 °C → −78 °C; 11, 1 h; −78 °C → −30 °C, 78%, dr = 5 : 1; (7) Ac2O, pyridine, 0 °C → rt; (8) DDQ, CH2Cl2–pH 7 buffer, 0 °C; (9) Dess–Martin periodinane, CH2Cl2, rt, 66% over three steps. | ||
The synthesis of the acylic precursor 4 was achieved by first alkylating aldehyde 7 with the Grignard reagent formed from alkyne 8. The diastereomeric mixture of alcohols was oxidized with Dess–Martin periodinane and the resulting ynone was converted into 4 by a double conjugate addition of 1,3-propanethiol. Treatment of 4 with dilute perchloric acid afforded a 1.00 : 0.74 : 0.26 mixture of spiroketals, which were determined to be the AB spiroketal core 5 and 2 spiroketals (9 and 6) that correspond to the CD-ring system. Separation of the spiroketals was achieved by preparative HPLC, and it was found that the undesired CD spiroketal system 9 could be equilibrated to the desired CD spiroketal 6 in 87% yield after three recycles. The two fragments 5 and 6 were transformed into the aldol-coupling fragments 10 and 11 in 13 and 10 steps respectively.
While the Evans group's anti-aldol coupling conditions were initially utilized,25 it was found that the reaction proceeded more efficiently when the solvent was diethyl ether rather than pentane, to yield the desired anti-aldol product in 65% yield. Functional group manipulation provided the aldehyde 2 required for the key Wittig coupling.
The EF fragment 3 was prepared from the acetal 12, which was synthesized from 3,4,5-tri-O-benzyl-D-glucal (Scheme 3).26 The acetal 12 was converted into the aldol precursor 13 in 18 steps. Formation of the boron enolate, followed by addition of the readily available aldehyde 14, afforded the aldol product 15 in 96% yield and with excellent stereocontrol due to the reaction proceeding under Felkin–Anh control. Removal of the TES protecting group induced cyclization, and the EF-ring system 16 was generated in 89% yield. Conversion of 16 into the required phosphonium salt 3 was achieved in 7 steps, with the critical C–C bond-forming step being a boron-mediated aldol reaction with aldehyde 17.
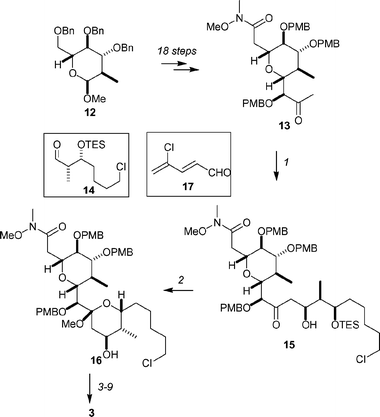 | ||
| Scheme 3 Ley's synthesis of the EF fragment 3 of spongistatin-1. Reagents and conditions: (1) Cy2BCl, NEt3, Et2O, 0 °C; 14, −78 °C, 96%; (2) PPTS, TMOF, MeOH, THF, rt, 89%. (3) TBSOTf, 2,6-lutidine, THF, −78 °C, 86%; (4) CeCl3, MeLi, THF, −78 °C, 96%; (5) Cy2BCl, NEt3, Et2O, 0 °C; 17, −78 °C; (6) TBSCl, imidazole, DMF, 97% over two steps; (7) Zn, PbI2, TMSCl, CH2I2, TiCl4, CH2Cl2–THF, rt, 75%; (8) NaI, nPrI, NaHCO3, Na2SO3, acetone, 93%; (9) PPh3, iPr2NEt, MeCN, 85 °C, quant. | ||
After considerable experimentation, the ylide was formed by reacting phosphonium salt 3 with MeLi·LiBr in THF (Scheme 4). Addition of the aldehyde 2 (which was dried with calcium hydride) resulted in the formation of the (Z)-alkene in 50% yield. Oxidative conditions removed all the PMB protecting groups, as well as hydrolyzing the methoxy ketal, to afford the (Z)-alkene 18. After deprotection of the allyl ester, the seco-acid intermediate was cyclized utilizing Evans’ modification of the Yamaguchi macrolactonization conditions. Global deprotection with HF afforded spongistatin-1, 1, in 57% yield.
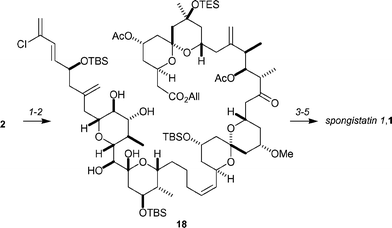 | ||
| Scheme 4 Completion of Ley's synthesis of spongistatin-1, 1. Reagents and conditions: (1) 3 treated with MeLi·LiBr, THF, −78 °C; then 2 (after drying with CaH2), −78 °C → rt, 50%; (2) DDQ, THF–CH2Cl2–pH 7 buffer, rt, 65%; (3) Pd(PPh3)4, morpholine, rt; (4) 2,4,6-trichlorobenzoyl chloride, iPr2NEt, DMAP, toluene, 90 °C, 56% over two steps; (5) 48% HF, MeCN, −21 °C, 57%. | ||
Full details of the Paterson synthesis of spongistatin-1 (1) have been reported in a series of articles.27
4 Polyethers
As noted in last year's review, structurally complex polyethers continue to draw significant attention from the synthesis community. Several reviews have been published in 2005,28 with Nakata, in particular, providing a major review of the total syntheses of marine polyethers.29Kadota, Yamamoto and co-workers have reported a convergent total synthesis of brevetoxin B, 19.30,31 This work utilizes a strategy for the synthesis of polycyclic ether frameworks whereby α-acetoxyethers are allylated in an intramolecular fashion, and cyclization is achieved by subsequent ring-closing metathesis.
The synthesis of the A→G fragment 20 is described in Scheme 5. Inspired by Hirama and Inoue's work on O,S-acetals for the synthesis of polycyclic ethers,32 the readily available BC and FG fragments 21 and 22 were coupled together by treatment with silver triflate to form the O,S-acetal 23. This material was readily converted into the allylic stannane 24 in 81% yield by a two-step procedure. First, 23 was reacted with γ-methoxyallylstannane in the presence of camphorsulfonic acid to form a methyl acetal, which was then selectively cleaved with TMSI/HMDS. Intramolecular allylation of 24 was found to proceed smoothly when carried out in the presence of silver triflate and provided a 78 : 22 mixture of the desired product 25 and its stereoisomer (not shown). After separation, the desired stereoisomer was transformed, in 18 steps, into the key A→G fragment 20.
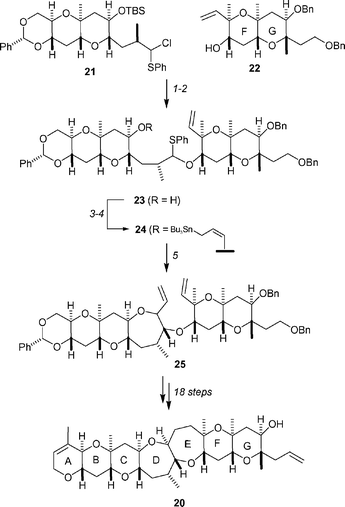 | ||
| Scheme 5 Kadota and Yamamoto's synthesis of the ABCDEFG fragment 20 of brevetoxin B, 19. Reagents and conditions: (1) AgOTf, DTBMP, 4 Å MS, CH2Cl2, −78 °C → −10 °C, 81% based on 22; (2) TBAF, THF, rt; (3) γ-methoxyallylstannane, CSA, CH2Cl2, rt, 81% over two steps; (4) TMSI, HMDS, CH2Cl2, 0 °C, 84%; (5) AgOTf, 4 Å MS, CH2Cl2, −78 °C → rt, 84% (dr = 78 : 22). | ||
Coupling of 20 with the JK-ring fragment 26 was achieved by esterification under Yamaguchi conditions to afford the ester 27 in 94% yield (Scheme 6). The ester 27 was converted to the allylic stannane 28 in a four-step procedure, which utilized the same protocol as discussed in Scheme 5. The α-chloroacetoxy ester was generated by a modified Rychnovsky acetylation of 27. Treatment of 28 with MgBr2·OEt2 in acetonitrile resulted in the formation of the ring-closing metathesis precursor 29 by an intramolecular allylation protocol. Ring-closing metathesis with Grubbs' first generation catalyst, followed by functional group manipulation, provided brevetoxin B, 19.
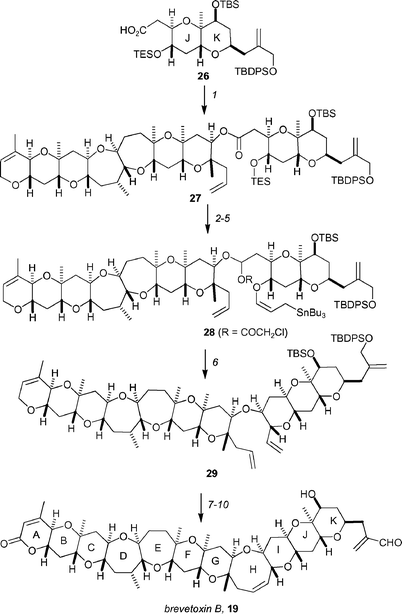 | ||
Scheme 6 Completion of Kadota and Yamamoto's synthesis of brevetoxin B, 19. Reagents and conditions: (1) 2,4,6-trichlorobenzoyl chloride, Et3N, THF, 40 °C, then 20, DMAP, toluene, rt, 94%; (2) TBAF, THF, 0 °C; (3) γ-methoxyallylstannane, CSA, CH2Cl2, rt; (4) HMDS, TMSI, CH2Cl2, 0 °C, 71%; (5) DIBAL-H, CH2Cl2, −78 °C, then (ClCH2CO)2O, DMAP, pyridine, CH2Cl2, −78 °C, 68%; (6) MgBr2·OEt2, MeCN, 40 °C, 82%; (7) (Cy3P)2Cl2Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh, benzene, 40 °C; (8) PCC, benzene, 80 °C, 81% for two steps; (9) HF·pyridine, CH2Cl2, 0 °C; (10) MnO2, Et2O, rt, 84% over two steps. CHPh, benzene, 40 °C; (8) PCC, benzene, 80 °C, 81% for two steps; (9) HF·pyridine, CH2Cl2, 0 °C; (10) MnO2, Et2O, rt, 84% over two steps. | ||
The Crimmins group has reported a stereoselective 22-step synthesis of the BCDE fragment of brevetoxin B, utilizing their established strategy for the synthesis of complex medium-ring ethers through a ring-closing metathesis of diene fragments.33
Rainier and co-workers have detailed a total synthesis of the ladder toxin gambierol,3430, that utilizes an iterative C-glycoside/enol ether–olefin ring-closing metathesis strategy to form the subunits (Scheme 7).35 Access to the key cyclization substrate 31 was achieved by coupling the ABC and FGH fragments 32 and 33 under Yamaguchi conditions. The original intent was to utilize the Takai–Utimoto titanium methylidene protocol to form the enol ether 34, which would be cyclized to form the E-ring system 35. However, the reaction between the olefin and the reagent formed from 1,1-dibromomethane, although preferentially giving the desired cyclic enol ether 35, was rather capricious. It was therefore surmized that using a titanium alkylidene generated from 1,1-dibromoethane would lead to a higher yield of cyclic enol ether 35. In the event, cyclic enol ether 35 was formed in 60% yield when ester 31 was reacted with the titanium alkylidene formed when 1,1-dibromoethane was used.
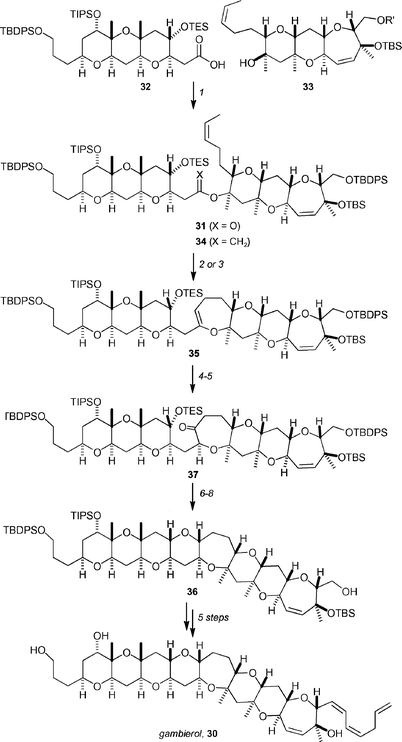 | ||
| Scheme 7 Key steps of Rainer's total synthesis of gambierol. Reagents and conditions: (1) 2,4,6-trichlorobenzoyl chloride, Et3N, THF, 40 °C; DMAP, toluene, 40 °C, 90%; (2) CH2Br2, Zn, PbCl2, TMEDA, TiCl4, CH2Cl2, THF, 30%; (3) CH3CHBr2, Zn, PbCl2, TMEDA, TiCl4, CH2Cl2, THF, 60%; (4) DMDO, CH2Cl2, −78 °C → 0 °C; DIBAL-H, CH2Cl2, 90%; (5) TPAP, NMO, 4 Å MS, CH2Cl2, rt, 97%; (6) CSA, MeOH, 0 °C, 90%; (7) Zn(OTf)2, EtSH, CH2Cl2, rt, 91%; (8) Ph3SnH, AIBN, toluene, 110 °C, 95%. | ||
Conversion of enol ether 35 to gambierol's octacyclic core 36 was achieved by first converting to the ketone 37 by a two-step procedure. Nicolaou's reductive cyclization protocol36 was used to form core structure 36 in 78% overall yield from 35. Completion of the synthesis was achieved by utilizing Yamamoto and Sasaki's protocols.37 Thus, gambierol was efficiently generated in 12 steps from the ABC and FGH subunits.
Mori and co-workers have completed a 20-step synthesis of the fully functionalized ABCD ring system of gambierol.38 They used their previously reported strategy for the construction of trans-fused polycyclic ether ring systems, which involves the alkylation of the oxiranyl anion generated from an α,β-epoxy sulfone followed by a 6-endo cyclization.39
A convergent synthesis of the CDEFG-ring system 38 of the gambieric acids40 has been described by Sato and Sasaki (Scheme 8).41 The CD fragment 39 and the G-ring fragment 40 were coupled under Yamaguchi conditions in 92% yield. This material was transformed into the alcohol 41 by application of the Rychnovsky reductive acetylation protocol, followed by generation of the nitrile group and deprotection with TBAF. Hydrolysis of the nitrile gave the carboxylic acid as a 1 : 1 mixture of isomers, which were converted into a mixture of seven-membered lactones by Yamaguchi lactonization. Unfortunately, the undesired isomer could not be isomerized to the desired lactone 42, but chromatography of the mixture provided the lactone 42 in 38% yield. Reductive acetylation, followed by stereoselective allylation with allyltrimethylsilane in the presence of BF3·OEt2, provided the diene in 67% yield. Formation of the nine-membered F-ring was achieved by ring-closing metathesis, using the second generation Grubbs' catalyst, to afford the CDEFG-ring system 38 in 98% yield.
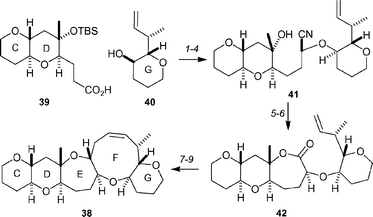 | ||
| Scheme 8 Sato and Sasaki's synthesis of the CDEFG-ring system 38 of the gambieric acids. Reagents and conditions: (1) 2,4,6-trichlorobenzoyl chloride, NEt3, DMAP, 92%; (2) DIBAL-H, −78 °C, then Ac2O, NEt3, DMAP, −78 °C → rt; (3) TMSCN, TMSOTf, DTBMP, −78 °C → 0 °C; (4) TBAF, THF, rt, 68% for three steps; (5) KOH, HOCH2CH2OH, 99%; (6) 2,4,6-trichlorobenzoyl chloride, NEt3, DMAP, 38% (+ 40% of epimer); (7) DIBAL-H, −78 °C, then Ac2O, NEt3, DMAP, −78 °C → 0 °C, 93%; (8) allyltrimethylsilane, BF3·OEt2, MeCN, −40 °C → 0 °C, 67%; (9) (H2IMes)(PCy3)Cl2RuCHPh, CH2Cl2, rt, 98%. | ||
Clark and co-workers have detailed a rapid two-directional synthesis of the FGHIJ fragment of the gambieric acids, which utilizes simultaneous double-ring closing metathesis reactions in an iterative manner.42
Full details of the Sasaki total synthesis of gymnocin A have been described.43 The convergent nature of the synthesis has allowed several analogs to be prepared and preliminary structure–activity relationship studies to be reported. The absolute configuration of gymnocin B has recently been determined.44
Tsukano and Sasaki have reported a synthetic approach to the NO-ring system 43 of gymnocin B (Scheme 9).45 The seven-membered O-ring containing 1,3-dimethyl groups presents a significant challenge due to steric congestion. After some experimentation, it was found that the NO-model system 43 could be prepared using ring-closing metathesis. Thus, diene 44 was generated from the known tetrahydropyranyl ketone 45 in 8 steps. The final step of this sequence involved the addition of vinyl magnesium bromide and led to the formation of a 1 : 1 mixture of diastereomers. Reaction with Grubbs' first generation catalyst afforded a 1 : 1 mixture of seven-membered ethers 46 and 47. Ether 46 was converted into the NO-model system 43 by hydrogenation of the alkene and deprotection. The other diastereomer 47 could be converted into 43 by a four-step sequence.
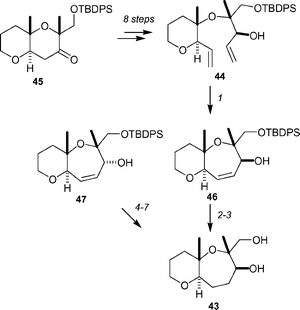 | ||
Scheme 9 The Tsukano–Sasaki approach to the NO-ring system 43 of gymnocin B. Reagents and conditions: (1) (PCy3)2Cl2RuCHPh, CH2Cl2, 35 °C, 40% for 46, 44% for 47; (2) H2, Pd/C, EtOAc, rt; (3) TBAF, THF, rt, 74% for two steps; (4) TPAP, NMO, 4 Å MS, CH2Cl2, 82%; (5) H2, Pd/C, EtOAc; (6) TBAF, THF, rt; (7) Me4N+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) BH(OAc)3−, MeCN–AcOH, −20 °C, 65% for three steps. BH(OAc)3−, MeCN–AcOH, −20 °C, 65% for three steps. | ||
Fujiwara and co-workers have detailed a convergent synthesis of the IJKLM-ring system 48 of ciguatoxin CTX3C (Scheme 10).46 This was achieved by first joining the I-ring fragment 49 to the L-ring fragment 50, then forming the K-ring using a reductive cyclization protocol. Alkylation of the dimethyldithioacetal 49 with 0.19 equivalents of the aldehyde 5047 produced a mixture of diastereomers, which were deprotected under acidic conditions. Ether 51 was then generated by cyclization of the hydroxyketone, using TMSOTf/Et3SiH, and protection of the primary hydroxyl group as a TBDPS ether. Functional group manipulation of 51 provided ketone 52, which upon reaction with TMSOTf/Et3SiH cyclized to form the J-ring, along with concomitant removal of the TBDPS ether. Oxidative radical cyclization provided a 1 : 1 mixture of spirocyclic acetal 48 and its C49 epimer. Equilibration of the mixture with camphorsulfonic acid in methanol provided the IJKLM-ring system 48 as the sole product in 73% yield.
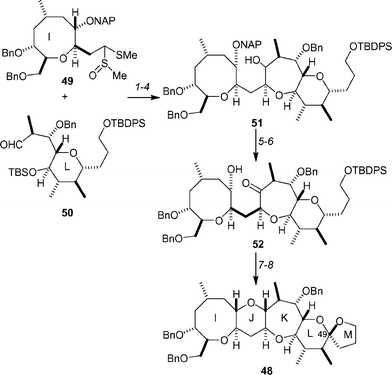 | ||
| Scheme 10 Fujiwara's synthesis of the IJKLM-ring system 48 of ciguatoxin CTX3C. Reagents and conditions: (1) 49, LDA, THF, −20 °C, 15 min, then 0.19 equiv. 50, −78 °C, 30 min, 98% from 50 (81% recovery of 49); (2) p-TsOH, MeOH, 23 °C, 30 min, 69%; (3) TMSOTf, Et3SiH, CH2Cl2, 0 °C, 30 min, 73%; (4) TBDPSCl, imidazole, DMF, 23 °C, 30 min., 87%; (5) (COCl)2, DMSO, NEt3, CH2Cl2, −78 °C, 10 min; (6) DDQ, CH2Cl2–pH 7 buffer (10 : 1), 23 °C, 10 min; (7) TMSOTf, Et3SiH, CH2Cl2, 0 °C, 30 min, 86% for 3 steps; (8) PhI(OAc)2, I2, hν, cyclohexane, 23 °C, 3 h, then CSA, MeOH, 23 °C, 5 h, 73%. | ||
The same group has also reported full details of their synthesis of the EF-ring segment of ciguatoxin CTX1B.48 Both Hirama and Isobe have recently summarized their efforts in the total synthesis of the ciguatoxins.49
McDonald and co-workers have extended their investigations into the biomimetic synthesis of trans,syn,trans-fused polyoxepanes and polypyrans by Lewis acid-initiated endo-regioselective tandem oxacyclization of polyepoxides.50 Giner has also investigated the formation of six-membered ether rings via an epoxyester–orthoester–cyclic ether rearrangement, which has been postulated as a possible biogenetic mechanism for the formation of ladder-type marine toxins.51
5 Batzelladines and crambescidins
Several syntheses of members of the marine guanadinium alkaloid family that includes that batzelladines and crambescidins have been reported in 2005. Gin and co-workers have described an elegant [4+2] annulation of carbodiimides that has resulted in a concise synthesis of batzelladine D, 63 (Scheme 11).52 The synthesis commenced with but-2-ynoate 53, which was treated with tetramethylguanidinium azide to afford the β-azido acrylate 54 in 84% yield (2 : 1 E : Z). Staudinger–aza-Wittig condensation with p-methoxybenzyl isocyanate provided vinyl carbodiimides (E)-55 and (Z)-55 with comparable efficiency, and set the stage for formation of the bicyclic dihydropyrimidine core. As such, reaction of either (E)- or (Z)-55 with 57 (readily prepared from lactam 56) produced 58 as a single diastereoisomer in 86% and 88% yield respectively. Desilylation (TBAF, 99%), followed by directed hydrogenation of the vinylogous carbamate (under Crabtree–Stork conditions, 80%) and subsequent IBX oxidation afforded the tricyclic guanidine hemiaminal 59 in 98% yield. The benzyl ester was removed with H2/Pd(OH)2 in 99% yield and was followed by Wittig olefination to give 60 (72%). Introduction of the guanidine-containing side chain by carboxylate alkylation with (BocHN)2CN(CH2)4OSO2Me yielded 61 (93%). The final ring was then formed by an iodoamination with I2 (70%) and reductive deiodination to give 62 in 89% yield. Removal of the protecting groups with TFA proceeded uneventfully in 82% yield, and secured a route to (−)-batzelladine D.
![The Gin synthesis of batzelladine D. Reagents and conditions: (1) (MeHN)2CNMe2+ N3−, CHCl3, 84% (2 : 1 E : Z); (2) (a) PPh3, CH2Cl2, 75% E, 58% Z, (b) p-MeOC6H4CH2NCO, PhMe, 85 °C, 73% E, 67% Z; (3) Cp2ZrHCl, THF, −20 °C, 66%; (4) 1,2-DCE, 23 °C, 86% from (E)-55, 88% from (Z)-55; (5) (a) TBAF, THF, 99%, (b) [Ir(cod)pyr(PCy3)]PF6, H2 (400 psi), CH2Cl2, 80%, (c) IBX, DMSO, CH3CN, 98%; (6) 10% Pd(OH)2/C, AcOH, H2 (1 atm), MeOH, 99%; (7) Me(CH2)8PPh3, THF, 50 °C, 72%; (8) (BocHN)2CN(CH2)4OMs, Cs2CO3, DMF, 40 °C, 93%; (9) I2, K2CO3, DME, 70%; (10) 10% Pd/C, Et3N, H2 (1 atm), EtOAc, 89%; (11) TFA, 82%.](/image/article/2007/NP/b602832m/b602832m-s11.gif) | ||
| Scheme 11 The Gin synthesis of batzelladine D. Reagents and conditions: (1) (MeHN)2CNMe2+N3−, CHCl3, 84% (2 : 1 E : Z); (2) (a) PPh3, CH2Cl2, 75% E, 58% Z, (b) p-MeOC6H4CH2NCO, PhMe, 85 °C, 73% E, 67% Z; (3) Cp2ZrHCl, THF, −20 °C, 66%; (4) 1,2-DCE, 23 °C, 86% from (E)-55, 88% from (Z)-55; (5) (a) TBAF, THF, 99%, (b) [Ir(cod)pyr(PCy3)]PF6, H2 (400 psi), CH2Cl2, 80%, (c) IBX, DMSO, CH3CN, 98%; (6) 10% Pd(OH)2/C, AcOH, H2 (1 atm), MeOH, 99%; (7) Me(CH2)8PPh3, THF, 50 °C, 72%; (8) (BocHN)2CN(CH2)4OMs, Cs2CO3, DMF, 40 °C, 93%; (9) I2, K2CO3, DME, 70%; (10) 10% Pd/C, Et3N, H2 (1 atm), EtOAc, 89%; (11) TFA, 82%. | ||
In other studies on the batzelladines, Nagasawa has provided details of their total syntheses of batzelladine A and batzelladine D.53 Synthetic material was used to identify their target protein, and it is concluded that batzelladines A and D bind specifically to CD4.
Overman has reported a total synthesis of the crambescidin core acid (Scheme 12) and has shown that the core acid readily decarboxylates to form crambescidin 359.54 Decarboxylation is fastest under basic conditions, and in the presence of base up to eight deuterium atoms can be incorporated. Both the deuterium incorporation and decarboxylation are results of the facile ring opening of the spirocyclic ether rings. The synthesis begins with urea 64 (available in 14 steps from 3-butynol), which was first converted to aminal 65 by dihydroxylation and then Pb(OAc)4 cleavage followed by addition of morpholinium acetate. Tethered Biginelli condensation with β-ketoester 6655 produced pyrrolopyrimidine 67 (along with the C13 epimer; dr = 6 : 1) in 63% yield over 3 steps from urea 64. Although the epimers could be separated by HPLC, they were carried forward as a mixture through a sequence that involved TBS and TIPS cleavage with TBAF in DMF to generate the diol, which underwent spirocyclization in the presence of 1 equivalent of p-TsOH at room temperature to form tricyclic urea 68 in 67% yield (over 2 steps). Protection of the alcohol as the α-chloroacetate and removal of the minor trans tricyclic isomer derived from 67 by silica gel chromatography provided 69 as a single diastereoisomer in 74% yield. Exposure of 69 to excess MeOTf in the presence of 2,6-di-tert-butyl-4-methylpyridine (DTBMP) yielded the expected intermediate pseudourea, which was subjected to anhydrous NH3 at 60 °C in allyl alcohol buffered with NH4Cl for 1.5 days to provide a 1.1 : 1.0 mixture of 70 and the C14 epimer, 71 in 78% yield. This mixture could be separated on silica gel, and 71 epimer could be equilibrated under the aminolysis conditions. After two recycles, pure 70 was obtained in 53% overall yield from tricyclic urea 69. Although a variety of palladium catalysts and reaction conditions30 were examined for the deprotection of cinnamate 70 to form core acid 72, 30 mol% of Pd(PPh3)4 at room temperature for 30 min with triethylammonium formate as the reducing agent proved to be optimal, and produced core acid as the zwitterion in high yield (>90% yield by 1H NMR analysis using an internal standard). Overman notes that purification of the core acid was challenging, as this zwitterion is a polar, highly crystalline solid, which is sparingly soluble in most solvents, and that it readily decarboxylates. Nonetheless, after some efforts, they were able to identify chromatography conditions that were effective for production of an analytically pure sample.
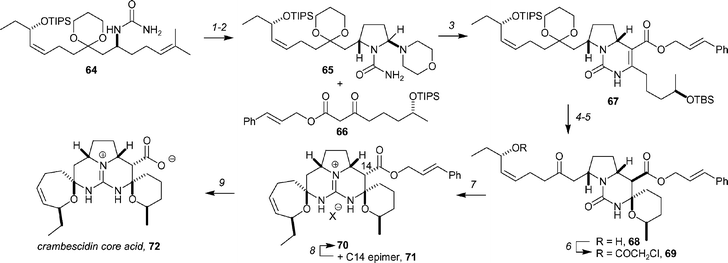 | ||
| Scheme 12 Overman's synthesis of the crambescidin core acid. Reagents and conditions: (1) 7.5 mol% OsO4, 3.5 equiv. NMO, THF, H2O; (2) Pb(OAc)4, PhMe then morpholinium acetate; (3) 66, CF3CH2OH, 60 °C, 63% (over 3 steps, dr = 6 : 1); (4) TBAF (5.5 equiv.), DMF; (5) TsOH·H2O (1 equiv.), CHCl3, rt, 67% (2 steps); (6) chloroacetyl chloride (5 equiv.), pyridine (20 equiv.), DMAP (0.5 equiv.), 74%; (7) MeOTf, DTBMP then NH3, NH4Cl, allyl alcohol, 60 °C, 78% (dr = 1.1 : 1); (8) NH3, NH4Cl, allyl alcohol, 60 °C, 53% combined yield of 70 after two recycles; (9) Pd(PPh3)4, Et3NH+HCO2−, THF, rt, 30 min, > 90%. | ||
The synthesis of crambidine has also been achieved by the Overman group (Scheme 13).56 Several approaches to the core of the molecule are described, but in the most concise route the synthesis commences with Biginelli adduct 75 (an intermediate in the Overman synthesis of 13,14,15-isocrambescidin 800).57 Desilylation with TBAF and cyclization with PPTS at 40 °C gave tetracycle 76 in 40% yield (2 steps). Dehydrogenation with CAN in acetonitrile then produced 77 in 65% yield. Palladium-catalyzed removal of the allyl ester (with morpholine as scavenger) proceeded in 75% yield, and set the stage for coupling of 78 with hydroxyspermidine derivatives 80 and 81. Coupling of acid 78 with the S enantiomer of Boc-protected hydroxyspermidine 81 gave amide (43S)-79 in 80% yield. Removal of the Boc groups from this compound with 3 M HCl at 0 °C provided (43S)-crambidine, 73, [α]D −18.3 (c 1.4, CH3OH), in 70% yield. Using the analogous procedure, acid 78 and 80 were converted to (43R)-crambidine, 74, [α]D −24.8 (c 0.25, CH3OH). 1H and 13C NMR spectra of these epimers were identical (as were the spectra for the peracetyl derivatives). Although the total synthesis was not able to unequivocally determine the stereochemistry at C43, Overman concludes that natural crambidine is most likely the 3S,8S,10S,19R,43S isomer 73.
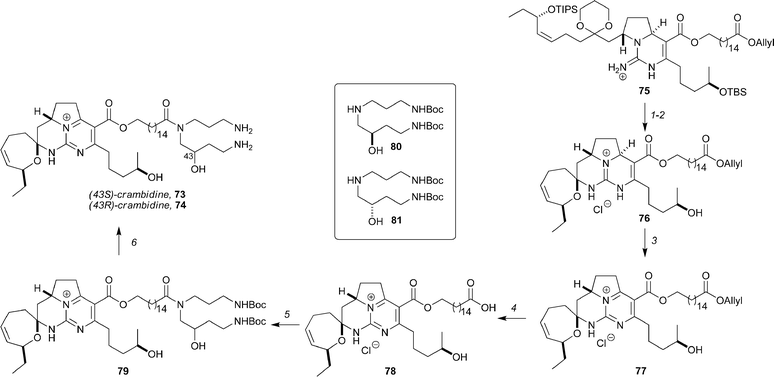 | ||
| Scheme 13 Overman's synthesis of crambidine. Reagents and conditions: (1) TBAF; (2) PPTS, 40 °C, 40% (2 steps); (3) CAN (3 equiv.), MeCN, NaHCO3 (10 equiv.), 65%; (4) Pd(PPh3)4, morpholine, rt, 75%; (5) 80 or 81, BOP, Et3N, ∼80%; (6) 3 M HCl, 0 °C, ∼75%. | ||
6 Amphidinolides
After a significant amount of activity on the synthesis front over the past 2 years, the amphidinolide family of polyketides saw only one new total synthesis in the review period: amphidinolide T4 from the Jamison group. In a similar vein to their earlier synthesis of amphidinolide T1, the synthesis employs a Ni-catalyzed reductive macrocyclization of an alkyne with an aldehyde (Scheme 14).58 Treating 82 with catalytic Ni(cod)2, Bu3P, and Et3B in toluene at 60 °C resulted in macrocyclization, and after protection of the alcohol and ozonolysis of the benzylidene unit, macrocycle 83 was obtained in 44% yield and with excellent diastereoselectivity (dr >10 : 1). Selective methylenation using a modification of Takai's method,59 and finally desilylation with HF·pyridine led to amphidinolide T4, 84 (74% over the final two steps). Jamison and Colby have also reviewed in a comparative fashion the synthesis of T-series amphidinolides,60 and described a route to the C13–C22 domain of amphidinolide T2.61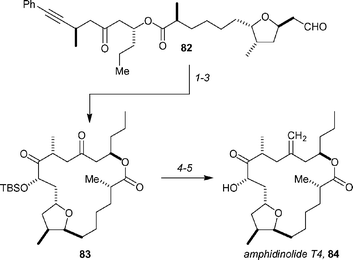 | ||
| Scheme 14 Key steps of Jamison's synthesis of amphidinolide T4. Reagents and conditions: (1) 20 mol% Ni(cod)2, 40 mol% Bu3P, Et3B, PhMe, 60 °C, (2) TBSCl, imidazole; (3) O3, then Me2S, >10 : 1 dr, 31% (3 steps); (4) CH2I2, Zn, ZrCl4, PbCl2; (5) HF·pyridine, 74% (2 steps). | ||
In other studies on the amphidinolides, a concise synthesis of the C9–C26 fragment of amphidinolide B162 has been accomplished by Carter and Zhang.63 The synthesis features a straightforward elimination approach to the C13–C15 diene. The Crews group has also described the synthesis of a number of subunits for amphidinolide B1 and an interesting approach to the C13–C15 diene based on coupling an allenic acetate with a B-alkyl borane.64 A convergent synthesis of the C9–C22 subunit of amphidinolide K in which both fragments start from 5-hydroxymethyltetrahydrofuran-2-one has been reported.65 An approach to amphidinolide H2 based on a Lewis acid-catalyzed Mukaiyama aldol reaction,66 a synthesis of the C6–C21 subunit of amphidinolide E,67 and a synthesis of the C1–C11 subunit of amphidinolide O that employs a 1,5-anti-selective aldol reaction have been reported.68 Trost has provided details of their elegant syntheses of amphidinolides P and A.69,70 A synthesis of a diastereoisomer of amphidinolide A has been reported.71
7 Dictyostatin-1 and discodermolide
Both discodermolide72 and dictyostatin-173 continue to attract substantial attention from the synthesis community, no doubt in large part due to their microtubule polymerization activities and the potential for either the parent compound or a related analog to be clinically relevant. The Panek group has reported a total synthesis of discodermolide, 85, that employs their chiral crotylsilane reagents for the construction of the C2–C4, C10–C12 and C16–C20 polypropionate-derived domains (Scheme 15). The synthesis commences with the preparation of key sub-units 86, 87, and 88 by the addition of chiral silanes to aldehydes. For example, addition of reagent A, under TiCl4 catalysis, to Roche ester derived aldehyde 89 gave 90 after the removal of the TBDPS group (85% for the two steps, dr >30 : 1). Subsequent PMP acetal formation and ozonolysis yielded 87. Application of the same general approach to the construction of 88 began with another TiCl4-catalyzed addition of silane B to aldehyde 91, to give 92 in 85% yield (dr >30 : 1). A sequence of 10 further steps provided 88. The synthesis of subunit 86 starts with the addition of silane C to aldehyde 89 (TiCl4, CH2Cl2, −78 °C), yielding 93 in 90% yield after the removal of the protecting groups. Introduction of a silylacetal and ozonolysis of the alkene gave 94, which was subjected to A in the presence of TiCl4 to give 95 (83% yield). This compound was advanced to 86 by a sequence of 11 steps.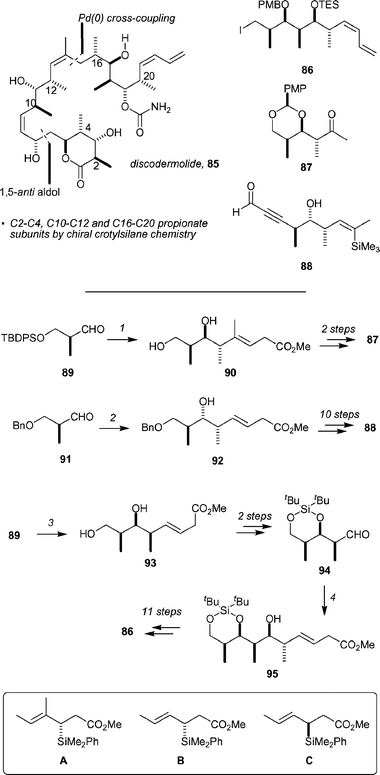 | ||
| Scheme 15 Key elements of the Panek synthesis of discodermolide, and the synthesis of subunits 86, 87, and 88 by crotylsilane chemistry. Reagents and conditions: (1) (a) A, TiCl4, CH2Cl2, −78 °C, (b) HCl, MeOH, 85%, dr >30 : 1; (2) B, TiCl4, CH2Cl2, −78 °C, 85%, dr >30 : 1; (3) (a) C, TiCl4, CH2Cl2, −78 °C to −30 °C, (b) HCl, MeOH, 90%, dr >30 : 1; (4) B, TiCl4, CH2Cl2, −78 °C to −30 °C, 83%, dr >30 : 1. | ||
The key subunit couplings and completion of the synthesis began with the addition of the dibutylboryl enolate of methyl ketone 87 to aldehyde 88 to yield 96 in an excellent 88% yield and with >30 : 1 diastereoselectivity (Scheme 16). Evans–Tischenko reduction of the β-hydroxyketone gave 97 in 95% yield. Subsequent hydrolysis of the β-hydroxybutyrate and exposure to mild acid gave 98 in 80% yield (the balance of material was saponified, but acetal migration had not occurred; this material could be converted to 98 in 80% yield by treatment with PPTS in CH2Cl2). A further 8 steps produced 99 and set the stage for the Pd-catalyzed cross-coupling to introduce the final fragment. This cross-coupling was possible with both borane nucleophiles such as 100 (generated from 86 by treatment with tBuLi and then 9-MeOBBN) in the presence of TlOEt and PdCl2dppf as catalyst, or zinc nucleophiles such as 101 (generated from 86 by treatment with tBuLi and then ZnCl2) in the presence of Pd(PPh3)4. The Suzuki approach yielded 82% of 102, and the Negishi approach yielded 64% of 102. In both cases the cross-coupling was sensitive to steric effects at C11 and failed when this alcohol was protected as a TBS ether. Discodermolide was then completed by a 5-step sequence consisting of protecting group removals and introduction of the carbamate. The overall sequence was 27 steps, and gave a 2.1% yield of discodermolide.
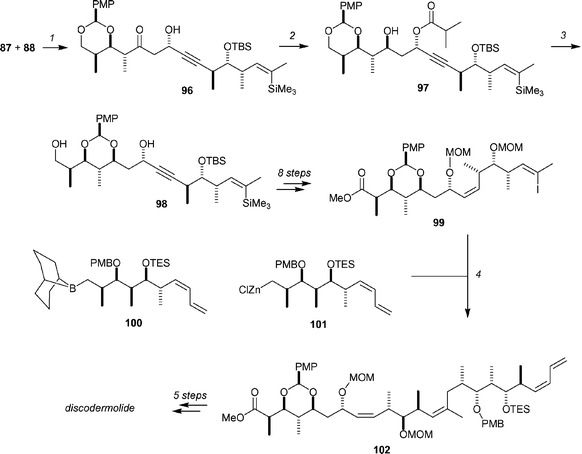 | ||
| Scheme 16 The final stages of the Panek synthesis of discodermolide. Reagents and conditions: (1) 87, Bu2BOTf, DIPEA, −115 °C, 88%, dr >30 : 1; (2) SmI2, iPrCHO, THF, −10 °C, 95%; (3) KOH/MeOH, then SiO2, 80%; (4) 100, PdCl2, dppf, TlOEt, DMF, 82% or101, Pd(PPh3)4, THF, 64%. | ||
Further studies from the Novartis group on the large-scale synthesis of discodermolide have resulted in a 24-step (longest linear sequence) formal synthesis of discodermolide that features improvements in several key reactions,74 and a detailed study of the key Paterson boron-based aldol reaction that was used for fragment coupling.75 A fourth-generation total synthesis with a longest linear sequence of 17 steps and an overall yield of 9.0% has been achieved by the Smith group at Penn.76 The Smith group has also communicated a number of papers in which synthesis has allowed the evaluation of a number of analogues including 14-normethyldiscodermolide,77 C13–C14 cyclopropane analogues and carbamate-substituted analogues.78 Studies of this type have revealed useful SAR information for a number of the functional groups in the molecule including the diene, the C7 hydroxy group and the lactone.79,80 Related work has resulted in the design and synthesis of fluorescent and photoaffinity molecular probes.81 The Paterson group has also reported the full details of some of their synthetic efforts that resulted in total syntheses.82
The Curran group has added two further papers on the heels of their total synthesis of dictyostatin-1: (a) a ring-closing metathesis route to the C10–C11 (Z)-alkene and (b) the synthesis and evaluation of 16-normethyldictyostatin.83 Synthetic 16-normethyldictyostatin had almost identical activity to the parent against cell-lines expressing wild-type tubulin, but variable activity against paclitaxel-resistant cell lines. Although not synthesis-centered, a paper describing the biological evaluations of dictyostatin that were facilitated by a synthetic route and details of the cellular effects of dictyostatin have been reported.84
8 Total syntheses of other compounds
Corey has described a strategy for the enantioselective synthesis of β-araneosene85 and isoedunol86 (members of the dolabellane family of marine natural products) that makes use of a number of nice ring-expansions (Scheme 17).87 Ester 103, which was ultimately derived from farnesol acetonide, was transformed to cyclopropanol 104 in 60% yield by a Kulinkovich reaction. Ring expansion to the cyclobutanone (104 → 105, 90%) was then achieved by treatment of 104 with Me3Al. Subsequent acidic hydrolysis of the acetonide, and periodate cleavage of the resulting diol gave keto aldehyde 106 in 92% yield, and set the stage for the reductive macrocyclization. After surveying a number of low-valent Ti-based reagents, it was discovered that slow addition (over 2 h by syringe pump) of 106 to a refluxing solution of SmI2 in THF provided the trans-pinacol product 107 as a single diastereomer in 78% yield. A sequence of 9 steps then yielded diol 108, the substrate for the second ring expansion. After optimization, the desired ring expansion to give 109 could be achieved in 98% yield by slow addition of Et3N to a solution of 108 and MsCl in CH2Cl2 at −30 °C, followed by slow warming to 4 °C and additional stirring for 72 h. The diastereoselective addition of 2-lithiopropene to ketone 109 provided an 82% yield of isoedunol, 110. Isoedunol could be transformed to β-aranosene, 111, by a two-step sequence consisting of MOM protection of the alcohol (92% yield) and reductive deoxygenation–alkene transposition under dissolving metal conditions (quantitative).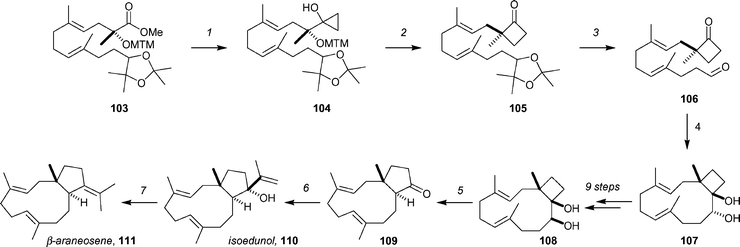 | ||
| Scheme 17 Key steps of the Corey syntheses of isoedunol and β-araneosene. Reagents and conditions: (1) 103, (iPrO)3TiCl (2.5 equiv.), THF, 0 °C, then EtMgBr (5 equiv), 60% (along with 35% of the ethyl ketone derived from 103); (2) Me3Al, CH2Cl2, −10 °C to 4 °C, 90%; (3) 4 : 1 AcOH–H2O, 55 °C, then NaIO4, 92%; (4) (a) syringe pump addition over 2 h of 106 to SmI2 (8 equiv.), THF, reflux, 78%, (b) Et3N, MsCl, CH2Cl2, 98%; (7) reverse addition of 109 to 2-lithiopropene, Et2O, −10 °C, 82%; (7) (a) MOMCl, iPr2NEt, TBAI, THF, 92%, (b) excess Li in NH3, THF, −60 °C, quantitative. | ||
Fukuyama has reported a concise synthesis of (−)-eudistomin C88 that employs an intramolecular Heck reaction and a highly diastereoselective Pictet–Spengler reaction (Scheme 18). Iodo-olefin 112, which was prepared in 8 steps from m-anisidine, was treated with Pd(OAc)2 to give indole 113 in 66% yield, which was then advanced to 114 by a sequence of 5 steps. Pictet–Spengler reaction of 114 with 115 in the presence of dichloroacetic acid in toluene (dr = 11 : 1) was followed by protection of the indole nitrogen as the α-chloroethoxycarbamate, to yield 116 in 89% yield over the two steps. Conversion of the MTM ether to the chloromethyl ether with SO2Cl2, followed by immediate reaction with AcSH in the presence of Hünig's base, provided 117 in 95% overall yield. Subsequent removal of the acetonide with aqueous acetic acid (61%), followed by mesylate formation (MsCl, 98%) furnished 118 and set the stage for the formation of the oxathiazepine ring. Upon heating with K2CO3 in MeOH, thiol acetate 118 underwent methanolysis, and subsequent cyclization produced the desired oxathiazepine ring, as well as removing the ACE group, in 65% yield. Final removal of the Boc group and phenolic methyl ether with BBr3 completed the synthesis of (−)-eudistomin C, 119, in 78% yield. Fukuyama and co-workers note that the 18-step sequence proceeds with an overall yield of 7.7%, and has allowed a gram-scale preparation of eudistomin C to be completed.
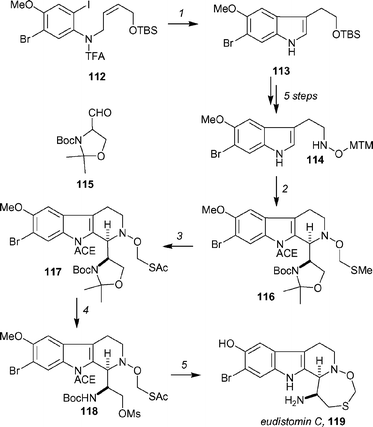 | ||
| Scheme 18 Fukuyama's synthesis of eudistomin C. Reagents and conditions: (1) cat. Pd(OAc)2, Et3N, BnEt3N+Cl−, DMF, 80 °C, 66%; (2) 115, cat. Cl2CHCO2H, toluene, 0 °C, dr = 11 : 1; (b) nBuLi, THF, −78 °C; ACE-Cl, 89% (two steps); (3) SO2Cl2, CH2Cl2, −78 °C → rt then AcSH, iPr2NEt, 95%; (4) (a) aq. AcOH, THF, 80 °C, 61%, (b) MsCl, iPr2NEt, CH2Cl2, 0 °C, 98%; (5) (a) K2CO3, MeOH, reflux, 65%, (b) BBr3, CH2CH2, −78 °C → rt, 78%. | ||
De Brabander and co-workers have completed a total synthesis of irciniastatin A,89120 (also known as psymberin90), which resolves the question of the C4 stereochemistry (this remained undetermined from structure elucidation).91 The synthesis is highly modular (Scheme 19, shown with the stereochemistry determined for 120), and should lend itself to further production of either the parent compound, or related structures. The assembly of subunits commences with an aldol reaction between the (Z)-chlorophenylboryl enolate obtained from ethyl ketone 121 (prepared in 8 steps from a known aldehyde) and aldehyde 122 (prepared in 7 steps from 2,4-dimethoxy-5-methylbenzaldehyde). This process yielded 123 in 88% yield and in high diastereoselectivity (dr = 12 : 1). A sequence of 5 steps then produced key amide 124, which was converted to the imidate with Meerwein's salt in the presence of polyvinylpyridine, which was readily removed by simple filtration after the completion of the reaction to yield the crude imidate 125. Acylation with the acid chloride derived from 126 (prepared with oxalyl chloride) in the presence of Hünig's base and subsequent reduction with NaBH4 yielded 127. After hydrolysis of the acetates, psymberin was obtained in 56% yield (from 124).
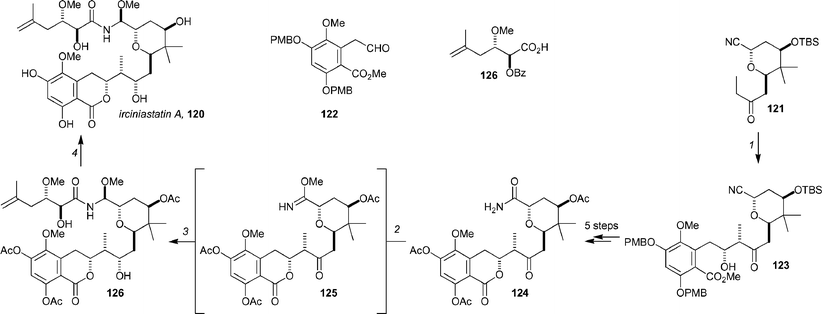 | ||
| Scheme 19 The key steps from De Brabander synthesis of psymberin/irciniastatin A. Reagents and conditions: (1) 121, PhBCl2, DIPEA, CH2Cl2, −78 °C, 88%, dr = 12 : 1; (2) Me3O+BF4−, polyvinylpyridine, CH2Cl2; filter; (3) (a) 126, (COCl)2, then add to 125, iPr2NEt, PhMe, 40 °C, (b) NaBH4, EtOH, 0 °C; (4) LiOH, MeOH, 56% (from 124). | ||
Marine-derived tetrahydroisoquinoline alkaloids have seen significant attention during the review period, and several total syntheses have been reported. The Magnus group and the Williams group simultaneously described total syntheses of renieramycin G92 (136, Scheme 20).93,94 The Williams approach to (−)-renieramycin G is based on the Williams synthesis of amino-acids, and begins with readily available lactam 128.95 A sequence of 4 steps provided 130, and treatment with ethyl glyoxylate afforded the corresponding anti-tetrahydroisoquinoline 131 as a single stereoisomer (92% yield). Advancement to 132 was achieved in 5 steps, and set the stage for coupling of the two domains. To this end, amino acid 129 (obtained from 128 in 6 steps) was converted into the acid chloride by treatment with oxalyl chloride, and was then coupled to 132 in the presence of 2,6-lutidine to yield 133 (89%). Selective removal of the primary TBS group, followed by oxidation under Swern conditions gave aldehyde 134 in excellent yield for two steps. Cyclization of compound 134 was accomplished in a one-pot procedure that involved initial treatment with TBAF to remove the phenolic O-TBS and N-Fmoc groups, producing an iminium intermediate through collapse of the resulting free secondary amine group onto the aldehyde group. Subsequent treatment with excess MsOH promoted nucleophilic attack by the electron-rich arene to give 135 in 89% yield. The synthesis was then completed by a straightforward route consisting of (1) removal of the O-allyl groups, (2) selective acylation of the primary alcohol group with angelic acid under modified Yamaguchi conditions (63% yield for two steps) and (3) DDQ oxidation of the hydroquinones to the bis-quinone (63% yield).
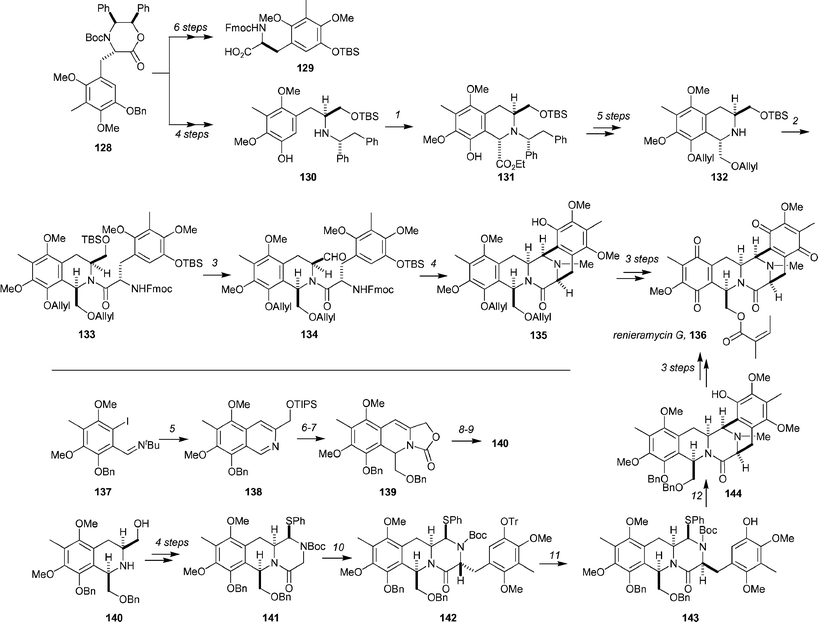 | ||
| Scheme 20 The Williams (top) and Magnus (bottom) syntheses of renieramycin G. Reagents and conditions: (1) EtO2CCHO, MeCN, 50 °C, 92%; (2) 129, (COCl)2, then 132, 2,6-lutidine, CH2Cl2, 89%; (3) HCO2H, THF, H2O, then Swern oxidation, 90%; (4) TBAF, 0 °C, then MsOH, 89%; (5) (a) triisopropyl(prop-2-ynyloxy)silane, CuI (1.2 equiv.), Et3N, 25 °C, 24 h, (b) CuI (0.2 equiv.), DMF, 100 °C, 1 h, 76%; (6) BnOCH2Li, THF, −78 °C, 2 h then ClCO2Me, 79%; (7) TBAF, THF, 0 °C, 91%; (8) TFA, Et3SiH, 0 °C; (9) H2NNH2, KOH, ethylene glycol, 150 °C, 86% (over 2 steps); (10) 2,4-dimethoxy-3-methyl-5-(trityloxy)benzyl chloride, KHMDS, 18-crown-6, THF, −78 °C, 20 min, 98%; (11) (a) tBuLi (2.2 equiv.), THF, −78 °C then BHT in THF, (b) HCl in Et2O, 0 °C, 1 h, 73% (over 2 steps); (12) (a) AgBF4, PhCl, 25 °C, 10 min, (b) CH2O, NaBH3CN, AcOH, MeOH, 25 °C, 65% (over 2 steps). | ||
The Magnus synthesis commences with o-iodoimine 137, which was subjected to a Castro coupling and a copper-catalyzed Larock isoquinoline synthesis to produce 138. Treatment of 138 with benzyloxymethyllithium, quenching with methyl chloroformate (79% yield), and removal of the TIPS ether (TBAF, 91%) gave oxazolidinone 139. Stereoselective reduction of the olefin with Et3SiH and TFA and cleavage of the oxazolidinone (H2NNH2, ethylene glycol, 150 °C gave the 1,3-cis-substituted tetrahydroisoquinoline 140. Four further steps led to 141. Subunit coupling was achieved by deprotonation of 141 with KHMDS and alkylation with the appropriate benzylic halide in the presence of 18-crown-6 to give 142 as a single diastereomer. Epimerization by deprotonation and diastereoselective reprotonation was employed to give 143 after trityl removal (dr = 6 : 1, separable). Phenol 143 was then cyclized by treatment with AgBF4. These conditions also resulted in Boc deprotection, and after reductive N-methylation, amine 33 was obtained. Completion of the synthesis was achieved by a sequence of 3 standard steps.
Danishefsky has reported a synthesis of cribrostatin IV that is based on the coupling of the two subunits 145 and 147 shown in Scheme 21.96 Compound 145 was prepared in 12 steps from commercially available 3-methyl-1,2-dimethoxybenzene, and compound 147 was prepared from 3-methylcatechol via compound 146. Compound 146 was subjected to Rh-catalyzed enantioselective reduction in the presence of (S,S)-Et-DuPhos, and subsequent ester hydrolysis and N-methylation provided 147. Subunit coupling between 145 and 147 was readily achieved in the presence of BOPCl and Et3N, to give 148 in 89% yield. Removal of the PMB group with DDQ (90% yield) and oxidation of both the primary and secondary alcohols gave 149 in 84% yield. Heating 149 to 100 °C in the presence of formic acid resulted in Mannich-type cyclization to give 150 in 59% yield. A sequence of 5 steps led to 151, and oxidation of the E-ring to the quinone with Fremy's salt (87%), followed by SeO2 oxidation, produced 152 in 84% yield. Oxidation of the benzylic alcohol (with Dess–Martin periodinane), and removal of the benzylic ether and conversion of the quinone to the hydroquinone gave 153 in 89% over the two steps. Angelation and removal of the TBS ether with TBAF/AcOH gave 154, which was advanced to cribrostatin IV, 155, in 65% yield by a sequence of three steps.
![The key steps of Danishefsky's synthesis of cribrostatin IV. Reagents and conditions: (1) Rh[(COD)–(S,S)-EtDuPhos]+ TfO−, H2 (100 psi), CH2Cl2–MeOH, 93%, 99% ee; (2) LiOH, MeOH–THF–H2O, 93%; (3) MeI, NaH, THF, 82%; (4) BOPCl, Et3N, CH2Cl2, 89%; (5) DDQ, CH2Cl2–pH 7 buffer, 90%; (6) Dess–Martin periodinane, 2,6-lutidine, CH2Cl2, 84%; (7) HCO2H, 100 °C, 59%; (8) (a) Fremy's salt, KH2PO4, CH3CN–H2O, 84%, (b) SeO2, dioxane, 100 °C, 87%; (9) DMP, CH2Cl2; (10) 10% Pd/C, H2 (1 atm), MeOH, 89% over two steps; (11) angeloyl chloride, CH2Cl2; (12) TBAF, AcOH, THF, 75% over two steps.](/image/article/2007/NP/b602832m/b602832m-s21.gif) | ||
| Scheme 21 The key steps of Danishefsky's synthesis of cribrostatin IV. Reagents and conditions: (1) Rh[(COD)–(S,S)-EtDuPhos]+TfO−, H2 (100 psi), CH2Cl2–MeOH, 93%, 99% ee; (2) LiOH, MeOH–THF–H2O, 93%; (3) MeI, NaH, THF, 82%; (4) BOPCl, Et3N, CH2Cl2, 89%; (5) DDQ, CH2Cl2–pH 7 buffer, 90%; (6) Dess–Martin periodinane, 2,6-lutidine, CH2Cl2, 84%; (7) HCO2H, 100 °C, 59%; (8) (a) Fremy's salt, KH2PO4, CH3CN–H2O, 84%, (b) SeO2, dioxane, 100 °C, 87%; (9) DMP, CH2Cl2; (10) 10% Pd/C, H2 (1 atm), MeOH, 89% over two steps; (11) angeloyl chloride, CH2Cl2; (12) TBAF, AcOH, THF, 75% over two steps. | ||
Baran and Richter have devised short enantioselective syntheses of welwitindolinone A (156) and fischerindoles I (157) and G (158, Scheme 22).97 This work is based an alternative biogenetic hypothesis for the conversion of 157 into 156 by an oxidative ring contraction. It was anticipated that 157 could be obtained by oxidation of 158. The synthesis of fischerindole G (158) was achieved by utilising the strategy previously developed by Baran and Richter for the synthesis of related fischerindoles.98 The required chloroketone 159 was readily generated by addition of vinyl magnesium bromide to the enolate of (R)-carvone oxide (160) to afford an alcohol 161, which was converted to the chloride 159 by reaction with NCS and PPh3. Coupling of ketone 159 with indole was achieved using the previously developed conditions to afford the indole 162 in 55% yield. The fischerindole ring system 163 was generated in 40% isolated yield (57% based on recovered starting material) by an intramolecular Friedel–Crafts cyclization of alkene 162. This cyclization was achieved by using Montmorillonite K-10 with microwave irradiation.
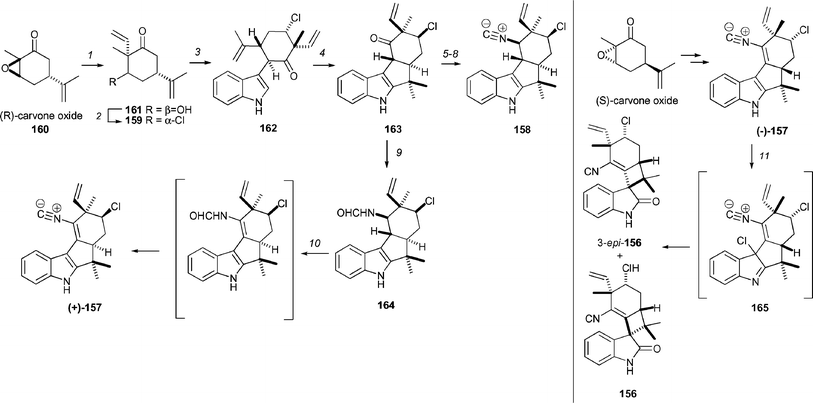 | ||
| Scheme 22 Baran's synthesis of welwitindolinone A (156) and fischerindoles I (157) and G (158). Reagents and conditions: (1) (a) LHMDS (1.2 equiv.), THF, −78 °C, 30 min, (b) −15 °C, CH2CHMgBr (2 equiv.), 15 min, 30%; (2) NCS (1 equiv.), PPh3 (1 equiv.), THF, 18 h, 55%; (3) indole (2 equiv.), THF, −78 °C, 30 min, then CuII2-ethylhexanoate (1.5 equiv.), −78 °C to 23 °C, 15 min, 55%; (4) Montmorillonite K-10 clay (10 equiv), DCE, microwave irradiation, 120 °C, 6 min, filter, then repeat, 40% + 30% recovered 7; (5) NaBH4, MeOH, 0 °C, 5 min; then Ms2O (2 equiv.), pyridine, rt, 30 min, 69%; (6) LiN3 (3 equiv.) DMF, 120 °C, 48 h, then Na–Hg (10 equiv.), EtOH, reflux, 4 h, 38%; (7) HCO2H (1.3 equiv.), CDMT (1.4 equiv.), DMAP (cat.), NMM (1.4 equiv.), CH2Cl2, 23 °C, 30 min, 87%; (8) Burgess reagent (2 equiv.), benzene, rt, 30 min, 82%; (9) NaBH3CN (10 equiv.), NH4OAc (40 equiv.), MeOH, THF, 7 days, 26% (48% brsm); (10) tBuOCl (1.5 equiv.), 0 °C, 10 min, then SiO2, Et3N, then Burgess reagent (2 equiv.), benzene, rt, 30 min, 47% overall; (11) tBuOCl (1.5 equiv.), −30 °C, 1 min, then 95 : 4 : 1 THF–H2O–TFA, −30 °C to 0 °C, 5 min, 28%, 10 : 1 mixture of 156 and 3-epi-156. | ||
While the original plan was to oxidize 158 to 157, it proved impossible to achieve this transformation. Addition of tBuOCl to 158 resulted in the formation of a stable C3-chloride species, which would require a difficult syn-elimination. Accordingly, it was decided to investigate whether elimination could occur when diastereomer 164 was used. This compound was generated by formylation of the amine that was obtained in 26% isolated yield (40% based on recovered starting material) by reductive amination of ketone 163. Treatment of formamide 164 with tBuOCl, followed by treatment with deactivated silica and then exposure to the Burgess reagent, resulted in the formation of fischerindole I [(+)-157] in 47% yield.
To synthesize welwitindolinone A (156) it was necessary to prepare the (−)-enantiomer of 157 using (S)-carvone oxide. Reaction of (−)-157 with freshly prepared tBuOCl at −30 °C generated 3-chloroindolenine 165, which upon dissolution in THF–H2O–TFA (95 : 4 : 1) and warming to 0 °C generated a 10 : 1 mixture of welwitindolinone A (156) and 3-epi-welwitindolinone A (3-epi-156) in 28% yield. The facile nature of this procedure would seem to indicate that this is a plausible biogenetic pathway.
Ningalin D99 (166, Scheme 23) is the most complex of the ningalin alkaloids and consists of a biphenylene quinone methide superimposed on an oxidized pentasubstituted pyrrole core. Boger and co-workers have completed a nine-step synthesis of ningalin D, which proceeds in 19% overall yield.100 The tetrasubstituted pyrrole 167 was rapidly assembled by firstly utilizing a inverse electron demand heterocyclic azadiene Diels–Alder reaction between symmetrical alkyne 168 and readily available tetrazine 169 to give symmetrical 1,2-diazine 170 in 87% yield. Reaction with 30 equivalents of zinc in trifluoroacetic acid at room temperature leads to cleavage of the diazine ring, followed by in situ cyclization to generate the pyrrole 167 in 64% yield. After alkylation of 167, the aryl C- and D-rings were formed by double Dieckmann condensation by reaction with NaH in DMF at room temperature. The resulting diphenol 171 was triflated and the aryl F- and G-rings attached by a double Suzuki coupling with boronic acid 172. Efforts to convert 173 into ningalin D were hampered by the steric congestion of the esters. However, hydrolysis of 173 with anhydrous hydroxide, followed by a modified Curtius rearrangement afforded permethyl ningalin D 174 in a remarkable 70% yield. Clearly, the expected diamine was oxidized in situ and the resulting imines were hydrolyzed upon workup to generate the desired biphenylene quinone methide system. The ten methyl ethers were removed by reaction of 174 with 15 equivalents of BBr3 to provide ningalin D, 166, in 96% yield.
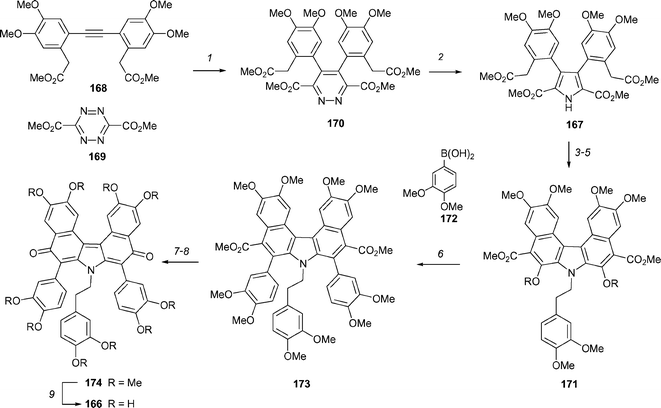 | ||
| Scheme 23 The Boger synthesis of ningalin B, 166. Reagents and conditions: (1) toluene, 110 °C, 87%; (2) Zn (30 equiv.), TFA, rt, 7 h, 64%; (3) (3,4-dimethoxyphenyl)ethyl iodide (3 equiv.), CsCO3 (3 equiv.), DMF, 60 °C, 2 h, 92%; (4) NaH (6 equiv.), DMF, 25 °C, 15 h, 81%; (5) Tf2O (5 equiv.), pyridine–CH2Cl2 (1 : 1), 0 °C → 25 °C, 2 h, 92%; (6) Pd(PPh3)4 (0.1 equiv), LiCl (2.2 equiv.), 1 M aq. K2CO3–DME, 80 °C, 15 h, 88%; (7) tBuOK (26 equiv.), H2O (8 equiv.), DMSO, 80 °C, 24 h, 84%; (8) (a) DPPA (5 equiv.), iPr2NEt (5 equiv.), CH2Cl2, 25 °C, 15–20 h, (b) H2O, THF, air, reflux, 90 h, 70%; (9) BBr3 (15 equiv.), CH2Cl2, −78 °C → rt, 16 h, 96%. | ||
The dictyodendrins101 are naturally occurring pyrrolo[2,3-c]carbazoles and have potential as telomerase inhibitors. Fürstner and co-workers have reported the first synthesis of dictyodendrin B, 175 (Scheme 24), which should allow further investigation of the biological properties of this unique class of natural product.102 The key transformations in this synthesis were the generation of a 2,3-disubstituted indole from a ketoamide using low-valent titanium and the subsequent 6π electrocyclization to form the pyrrolcarbazole ring system. The required ketoamide 176 was readily available from coupling of acid chloride 177 with the amine 178. Exposure of ketoamide 176 to titanium/graphite in the presence of pyridine generated the indole 179 in 93% yield. Irradiation of 179 with UV light initiated the 6π electrocyclization and the product was aromatized in situ by reaction with Pd/C and nitrobenzene to afford the pyrrolocarbazole 180 in 81% yield. After some experimentation, it was found that the best way to complete the carbon skeleton was by a two-step protocol. Bromination of 180 with NBS, followed by halogen–metal exchange, generated an aryllithium species, which was treated with 4-methoxybenzaldehyde to form the secondary alcohol 181. It was found that the oxidation of 181 was problematic due to the generation of an unsymmetrical dimer (not shown). However, TPAP/NMO oxidation under dilute conditions (0.01 M) generated the ketone in 66% yield. Selective cleavage of the isopropyl ether with BCl3 allowed for the introduction of the sulfate group by reaction of the resulting phenol with trichloroethyl chlorosulfonic acid ester. Exhaustive demethylation with BCl3/Bu4NI, followed by reductive cleavage of the chloroethyl ester, afforded dictyodendrin B, 175, in 58% yield. The longest linear sequence is 13 steps and the synthesis proceeded in 8% overall yield.
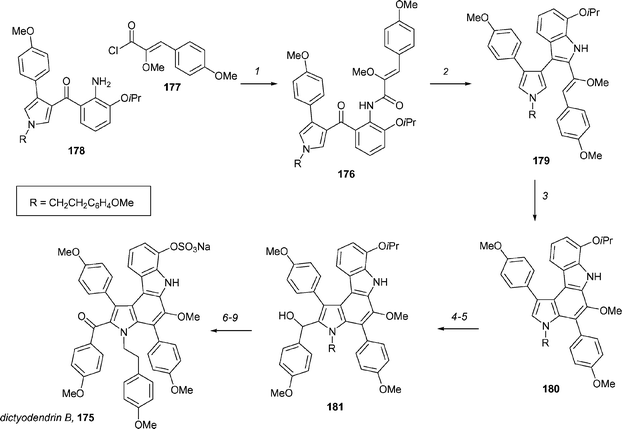 | ||
| Scheme 24 The Fürstner synthesis of dictyodendrin B. Reagents and conditions: (1) 177, CH2Cl2, Et3N, cat. DMAP, 89%; (2) TiCl3–2KC8, DME, pyridine, reflux, 71–93%; (3) hν, MeCN, cat. Pd/C, PhNO2, 81%; (4) NBS, THF, 0 °C, 69%; (5) (a) MeLi, THF, −78 °C, (b) nBuLi, −78 °C; (c) p-MeOC6H4CHO, −78 °C → rt, 97%; (6) TPAP (10%), NMO, 4 Å MS, CH2Cl2 (0.01 M), 66%; (7) BCl3, CH2Cl2, −20 °C, 85%; (8) Cl3CCH2OSO2Cl, DABCO, CH2Cl2, 92%; (9) (a) BCl3, TBAI, CH2Cl2, 0 °C, rt, (b) Zn, HCO2−NH4+, MeOH, 58%. | ||
A large number of other total synthesis of marine natural products were reported in the review period, and papers describing first total syntheses are presented in Table 1. New total syntheses of compounds previously prepared are summarized in Table 2.
| Compound | Reference | Notes |
|---|---|---|
(−)-Isoprelaurefucin
|
Kim et al.104 | • 14 Steps from known compound |
| • Non-racemic | ||
| • Bioactivity: unknown | ||
(−)-Siphonodiol and (−)-tetrahydrosiphonodiol
|
López et al.105 | • Each 9 steps from L-gulono-1,4-lactone |
| • Non-racemic | ||
| • Bioactivity: antifungal, antibacterial, metamorphosis-inducing activity and antifouling | ||
Luffolide and 2 related sesterterpenolides
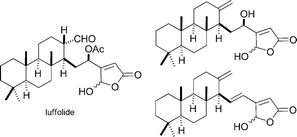
|
Basabe et al.106 | • Luffolide: 12 steps from methyl isoanticopalate |
| • Non-racemic | ||
| • Bioactivity: anti-inflammatory | ||
(−)-Isodomoic acid C
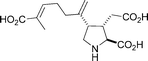
|
Clayden et al.107 | • 17 Steps from cumylamine |
| • Non-racemic | ||
| • Bioactivity: insecticidal and implicated in amnesic shellfish poisoning | ||
Placidenes
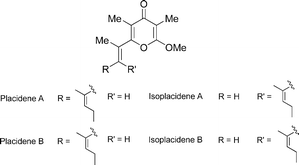
|
Trauner et al.108 | • Each 5 steps from known compound |
| • Bioactivity: potential sunscreens | ||
Psammaplysenes A and B

|
Georgiades and Clardy109 | • Psammaplysene A: 8 steps from 4-iodophenol |
| • Psammaplysene B: 9 steps from 4-iodophenol | ||
| • Bioactivity: inhibitors of FOXO1a-mediated nuclear export. | ||
Pachastrissamine (jaspine B)
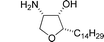
|
Datta et al.110 and Rao et al.111 | • 14 Steps from L-serine (Datta) |
| • 10 Steps from Garner's aldehyde (Rao) | ||
| • Non-racemic | ||
| • Bioactivity: cytotoxic against several human cancer cell lines | ||
Putative structure of palau'amide
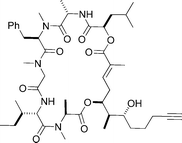
|
Ma et al.112 | • Data doesn't match that reported for the natural product. |
| • 19 Steps from 5-hexynal | ||
| • Non-racemic | ||
| • Bioactivity: potent cytotoxicity | ||
Putative structure of tridachiahydropyrone
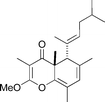
|
Perkins et al.113 | • Data doesn't match that reported for the natural product. |
| • 12 Steps from known compound | ||
| • Non-racemic | ||
| • Bioactivity: unknown | ||
Methyl sarcoate
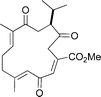
|
Nakata et al.114 | • 16 Steps from known compound |
| • Non-racemic | ||
| • Bioactivity: unknown | ||
Pelorol
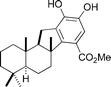
|
Anderson et al.115 | • 11 Steps from (+)-sclareolide |
| • Non-racemic | ||
| • Bioactivity: weak antimicrobial activity | ||
Yanuthones
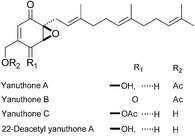
|
Mehta and Chandra Pan116 | • Yanuthone C and 22-deacetylyanuthone A: 7 steps from 4-methoxyphenol |
| • Yanuthones A and B: 8 steps from 4-methoxyphenol | ||
| • Racemic | ||
| • Bioactivity: inositol-5-phosphatase SHIP activator | ||
Symbioramide
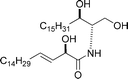
|
Sugai et al.117 | • 12 Steps from known compound |
| • Chemo-enzymatic resolution | ||
| • Bioactivity: Ca2+-ATPase activating factor, antileukemic substance against L1210 murine leukemia cells | ||
Semiplenamide C
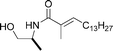
|
Bull et al.118 | • 5 Steps from L-alanine methyl ester |
| • Non-racemic | ||
| • Bioactivity: toxicity toward the brine shrimp model system | ||
Phakellistatins 7–9
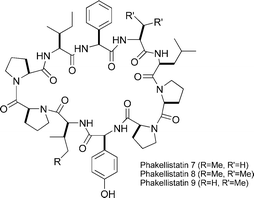
|
Gomez-Paloma et al.119 | • 24 Steps |
| • Non-racemic | ||
| • Bioactivity: in vitro cytotoxic activity against a panel of cancer cell lines. | ||
(+)-Austrodoral
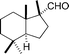
|
Alvarez-Manzaneda et al.120 and Gavagnin et al.121 | • 8 Steps from (−)-sclareol (Alvarez-Manzaneda) |
| • 8 Steps from (+)-sclareolide (Gavagnin) | ||
| • Non-racemic | ||
| • Bioactivity: ichthyotoxic | ||
Agardhilactone

|
Miyaoka et al.122 | • Assignment of absolute configuration |
| • 17 Steps from known compound | ||
| • Non-racemic | ||
| • Bioactivity: unknown | ||
Calafianin
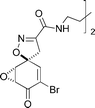
|
Ogamino and Nishiyama123 | • Structural revision |
| • 6 Steps from known compound | ||
| • Racemic | ||
| • No significant bioactivity | ||
(−)-Aurisides A and B
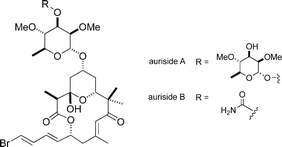
|
Paterson et al.124 | • Auriside A: 18 steps from known compound |
| • Auriside B: 17 steps from known compound | ||
| • Non-racemic | ||
| • Bioactivity: cytotoxic | ||
Hallipeptin A
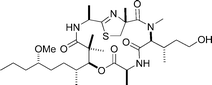
|
Ma et al.125 | • 21 Steps from known compound |
| • Non-racemic | ||
| • Bioactivity: potent inflammatory activity | ||
Hallipeptin D
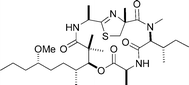
|
Nicolaou et al.126 | • 24 Steps from known compound |
| • Non-racemic | ||
| • Bioactivity: cytotoxic | ||
Curcutetraol
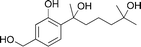
|
Lindel et al.127 | • 7 Steps from known compound |
| • Racemic | ||
| • Bioactivity: unknown | ||
(−)-Marginatone
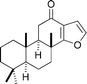
|
Ragoussis et al.128 | • 3 Steps from (+)-coronarin |
| • Non-racemic | ||
| • Bioactivity: unknown | ||
(−)-Antipode of cis-dihydrohamacanthin B
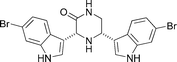
|
Kawasaki et al.129 | • 14 Steps from known compound |
| • Non-racemic | ||
| • Bioactivity: a number of these metabolites were shown to possess antifungal, antiinflammatory, antitumor, antiviral and cytotoxic activities, and inhibition of serine-threonine protein phosphatase and neural nitric synthetase. | ||
Bastadins 5, 10, 12, 16, 20, and 21
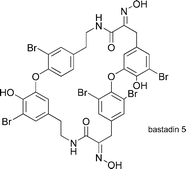
|
Couladouros et al.130 | • 22–23 Steps from 4-bromophenol |
| • Bioactivity: anticancer, antimicrobial activity, inhibition of inosine-5′-phosphate dehydrogenase, inhibition of lipoxygenases, and specific interaction with intracellular calcium channels. | ||
A thiomarinol antibiotic
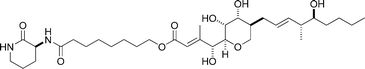
|
Gao and Hal131 | • 17 Steps |
| • Non-racemic | ||
| • Bioactivity: anti-bacterial | ||
3-Bromobarekoxide and laukarlaol
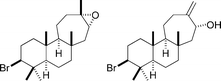
|
Justica et al.132 | • 6 and 7 Steps respectively |
| • Racemic | ||
| • Bioactivity: unknown | ||
| • Structure of laukarlaol revised to that shown | ||
(+)-Bistramide C
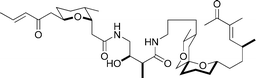
|
Wipf and Hopkins133 | • 31 Steps |
| • Non-racemic | ||
| • Bioactivity: antiproliferative, sodium channel blockage, protein kinase Cδ activation | ||
(+)-Cyanthiwigin U
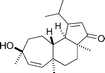
|
Pfeiffer and Phillips134 | • 12 Steps |
| • Non-racemic | ||
| • Bioactivity: members of the cyanthiwigin class are cytotoxic and have activity against M. tuberculosis |
| Compound | Reference | Compound | Reference |
|---|---|---|---|
| Pseudopteroxazole | Harmata and Hong135 | Manzacidin C | Lanter et al.136 |
| Cyercene A | Trauner et al.137 | (−)-Curcuquinone | Minatti and Dötz138 |
| Clasto-lactacystin β-lactone | Donohoe et al.139 | (±)-Mycothiazole | Cossy et al.140 |
| (−)-Dactylolide | Ding and Jennings141 | (−)-Agelastatin A | Davis and Deng142 |
| Mahafacyclin B | Jolliffe et al.143 | Cryptophycin-1 | Sewald et al.144 |
| Octalactin A | Shiina et al.145 | Taurospongin A | Campagne et al.146 |
| Hallipeptin A | Nicolaou et al.147 | Bengamide E | Sarabia and Sanchez-Ruiz148 |
| (−)- and (+)-cis-Lauthisan | Solladié et al.149 | (±)-Dibromophakellstatin | Lindel et al.150 |
| (±)-Flustramines A and C | Fuchs and Funk151 | Tanikolide | Zhai et al.152 |
| (R)-Strongylodiols A and B | Baldwin et al.153 | (−)-Harzialactone A | Wu et al.154 |
| Hamigeran B | Trost et al.155 | Bastadin 6 | Kobayashi et al.156 |
| Eleutherobin | Gennari et al.157 | (+)-Erogorgiaene | Davies and Walji158 |
| Polycitones A and B | Gupton et al.159 | (−)-Laulimalide | Uenishi and Ohmi160 |
| Desmethylubiquinone Q2 | Stonik et al.161 | (±)-Herbindole B and (±)-cis-trikentrin B | Kerr et al.162 |
| (±)-Pinnaic Acid and (±)-halichlorine | Andrade and Martin163 | (−)-Colombiasin A and (−)-elisapterosin B | Harrowven et al.164 |
| (−)-Dactylolide and (−)-zampanolide | Ding and Jennings165 | (−)-Kainic acid | Fukuyama et al.166 and Greene et al.167 |
| (±)-9-Isocyanoneopupukeanane | Srikrishna and Satyanarayana168 | Dibromotyrosine alkaloid inhibitor of mycothiol-S-conjugate amidase | Chanda and Sulake169 |
| (Z)-Hymenialdisine and (Z)-2-debromohymenialdisine | Papeo et al.170 | trans-Dragmacidin C, and cis- and trans-dihydrohamacanthin A | Garg and Stoltz171 |
| (+)-Laurencin | Fujiwara et al.172 and Kim et al.173 | (+)-Cylindricine C and (−)-fasicularin | Kibayashi et al.174 |
| (+)-Peloruside A | Jin and Taylor175 | Lamellarin D | Alvarez et al.176 |
| (+)-Leucascandrolide A | Su and Panek177 |
9 Acknowledgements
We would like to thank Professor John Blunt and Professor Murray Munro (University of Canterbury, Christchurch, New Zealand) for a copy of the 2006 version of the MarinLit database,103 which facilitated data collection for this review.10 References
- J. W. Blunt, B. R. Copp, W.-P. Hu, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2007, 24 10.1039/b603047p.
- K.-S. Yeung and I. Paterson, Chem. Rev., 2005, 105, 4237 CrossRef CAS.
- J. Hassfeld, M. Kalesse, T. Stellfeld and M. Christmann, Adv. Biochem. Eng. Biotechnol., 2005, 97, 133 Search PubMed.
- D. E. N. Jacquot and T. Lindel, Curr. Org. Chem., 2005, 9, 1551 CrossRef CAS.
- M. Inoue, Chem. Rev., 2005, 105, 4379 CrossRef CAS.
- C. Kibayashi, Chem. Pharm. Bull., 2005, 53, 1375 CrossRef CAS.
- M. J. Yu, Y. Kishi and B. A. Littlefield, in Anticancer Agents from Natural Products, ed. G. M. Cragg, D. G. I. Kingston and D. J. Newman, CRC Press, Boca Raton, 2005, pp. 241–266 Search PubMed.
- R. Henriquez, G. Faircloth and C. Cuevas, in Anticancer Agents from Natural Products, ed. G. M. Cragg, D. G. I. Kingston and D. J. Newman, CRC Press. Boca Raton, 2005, pp. 215–240 Search PubMed.
- K. C. Nicolaou, J. Org. Chem., 2005, 70, 7007 CrossRef CAS.
- D. Sipkema, R. Osinga, W. Schatton, D. Mendola, J. Tramper and R. H. Wijffels, Mar. Biotechnol., 2005, 90, 201 CAS.
- J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2005, 22, 15 RSC.
- T. L. Simmons, E. Andrianasolo, K. McPhail, P. Flatt and W. H. Gerwick, Mol. Cancer Ther., 2005, 4, 333 Search PubMed.
- T. J. Heckrodt and J. Mulzer, Top. Curr. Chem., 2005, 244, 1 CAS.
- V. M. Dembitsky, T. A. Gloriozova and V. V. Poroikov, Mini-Rev. Med. Chem., 2005, 5, 319 Search PubMed.
- N. T. Patil and Y. Yamamoto, Recent Res. Dev. Org. Chem., 2005, 9, 131 Search PubMed.
- M. Hiersemann and H. Helmboldt, Top. Curr. Chem., 2005, 243, 73 CAS.
- Y. Hamada and T. Shioiri, Chem. Rev., 2005, 105, 4441 CrossRef CAS.
- B. Stanovnik and J. Svete, Mini-Rev. Org. Chem., 2005, 2, 211 Search PubMed.
- D. L. J. Clive, M. Yu, J. Wang, V. S. C. Yeh and S. Kang, Chem. Rev., 2005, 105, 4483 CrossRef CAS.
- N. Ungur and V. Kulcitki, Phytochem. Rev., 2005, 3, 401 Search PubMed.
- K.-I. Takao, R. Munakata and K.-I. Tadano, Chem. Rev., 2005, 105, 4779 CrossRef CAS.
- E. J. Kang and E. Lee, Chem. Rev., 2005, 105, 4348 CrossRef CAS.
- G. R. Pettit, Z. A. Chicacz, F. Gao, C. L. Herald, M. R. Boyd, J. M. Schmidt and J. N. A. Hooper, J. Org. Chem., 1993, 58, 1302 CrossRef CAS.
- (a) M. J. Gaunt, A. S. Jessiman, P. Orsini, H. R. Tanner, D. F. Hook and S. V. Ley, Org. Lett., 2003, 5, 4819 CrossRef CAS; (b) M. J. Gaunt, D. F. Hook, H. R. Tanner and S. V. Ley, Org. Lett., 2003, 5, 4815 CrossRef CAS.
- D. A. Evans, P. J. Coleman and L. C. Dias, Angew. Chem., Int. Ed. Engl., 1985, 36, 2738.
- S. Kawahara, M. J. Gaunt, A. Scolaro, S. Yamanoi and S. V. Ley, Synlett, 2005, 2031 CAS.
- (a) I. Paterson, M. J. Coster, D. Y.-K. Chen, R. M. Oballa, D. J. Wallace and R. D. Norcross, Org. Biomol. Chem., 2005, 3, 2399 RSC; (b) I. Paterson, M. J. Coster, D. Y.-K. Chen, K. R. Gibson and D. J. Wallace, Org. Biomol. Chem., 2005, 3, 2410 RSC; (c) I. Paterson, M. J. Coster, D. Y.-K. Chen, J. L. Acena, J. Bach, L. E. Keown and T. Trieselmann, Org. Biomol. Chem., 2005, 3, 2420 RSC; (d) I. Paterson, D. Y.-K. Chen, M. J. Coster, J. L. Acena, J. Bach and D. J. Wallace, Org. Biomol. Chem., 2005, 3, 2431 RSC.
- (a) I. Kadota and Y. Yamamoto, Acc. Chem. Res., 2005, 38, 423 CrossRef CAS; (b) L. Hintermann, Nachr. Chem., 2005, 53, 646 Search PubMed.
- T. Nakata, Chem. Rev., 2005, 105, 4314 CrossRef CAS.
- I. Kadota, H. Takamura, H. Nishii and Y. Yamamoto, J. Am. Chem. Soc., 2005, 127, 9246 CrossRef CAS.
- Y.-Y. Lin, M. Risk, S. M. Ray, D. Van Engen, J. Clardy, J. Golik, J. C. James and K. Nakanishi, J. Am. Chem. Soc., 1981, 103, 6773 CrossRef CAS.
- (a) M. Inoue, G. X. Wang, J. Wang and M. Hirama, Org. Lett., 2002, 4, 3439 CrossRef CAS; (b) M. Inoue, K. Miyazaki, H. Uehara, M. Maruyama and M. Hirama, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12013 CrossRef CAS.
- M. T. Crimmins, P. J. McDougall and K. A. Emmitte, Org. Lett., 2005, 7, 4033 CrossRef CAS.
- M. Satake, M. Murata and T. Yasumoto, J. Am. Chem. Soc., 1993, 115, 361 CrossRef CAS.
- H. W. B. Johnson, U. Majumder and J. D. Rainier, J. Am. Chem. Soc., 2005, 127, 848 CrossRef CAS.
- K. C. Nicolaou, C. V. C. Prasad, C. K. Hwang, M. E. Duggan and C. A. Veale, J. Am. Chem. Soc., 1989, 111, 5321 CrossRef CAS.
- (a) H. Fuwa, M. Sasaki, M. Satake and K. Tachibana, Org. Lett., 2002, 4, 2981 CrossRef CAS; (b) H. Fuwa, N. Kainuma, K. Tachibana and M. Sasaki, J. Am. Chem. Soc., 2002, 124, 14983 CrossRef CAS; (c) I. Kadota, H. Takamura, K. Sato, A. Ohno, K. Matsuda and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 46 CrossRef CAS; (d) I. Kadota, H. Takamura, K. Sato, A. Ohno, K. Matsuda, M. Satake and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 11893 CrossRef CAS; (e) I. Kadota, A. Ohno, Y. Matsukawa and Y. Yamamoto, Tetrahedron Lett., 1998, 39, 6373 CrossRef CAS.
- H. Furuta, M. Hase, R. Noyori and Y. Mori, Org. Lett., 2005, 7, 4061 CrossRef CAS.
- (a) Y. Mori, K. Yaegashi, K. Iwase, Y. Yamamori and H. Furukawa, Tetrahedron Lett., 1996, 37, 2605 CrossRef CAS; (b) Y. Mori, K. Yaegashi and H. Furukawa, J. Am. Chem. Soc., 1996, 118, 8158 CrossRef CAS.
- H. Nagai, K. Torigoe, M. Satake, M. Murata, T. Yasumoto and H. Hirota, J. Am. Chem. Soc., 1992, 114, 1102 CrossRef CAS.
- K. Sato and M. Sasaki, Org. Lett., 2005, 7, 2441 CrossRef CAS.
- J. S. Clark, M. C. Kimber, J. Robertson, C. S. P. McErlean and C. Wilson, Angew. Chem., Int. Ed., 2005, 44, 6157 CrossRef CAS.
- C. Tsukano, M. Ebine and M. Sasaki, J. Am. Chem. Soc., 2005, 127, 4326 CrossRef CAS.
- K. Tanaka, Y. Itagaki, M. Satake, H. Naoki, T. Yasumoto, K. Nakanishi and N. Berova, J. Am. Chem. Soc., 2005, 127, 9561 CrossRef.
- C. Tsukano and M. Sasaki, Tetrahedron Lett., 2005, 46, 4617 CrossRef CAS.
- D. Domon, K. Fujiwara, A. Murai, H. Kawai and T. Suzuki, Tetrahedron Lett., 2005, 46, 8285 CrossRef CAS.
- D. Domon, K. Fujiwara, Y. Ohtaniuchi, A. Takezawa, S. Takeda, H. Kawasaki, A. Murai, H. Kawai and T. Suzuki, Tetrahedron Lett., 2005, 46, 8279 CrossRef CAS.
- A. Takemura, K. Fujiwara, K. Shimawaki, A. Murai, H. Kawai and T. Suzuki, Tetrahedron, 2005, 61, 7392 CrossRef CAS.
- (a) M. Isobe, S. Takai, T. Baba and Y. Hamashima, Gendai Kagaku Zokan, 2005, 43, 181 Search PubMed; (b) M. Hirama, Chem. Rec., 2005, 5, 240 CrossRef CAS.
- J. C. Valentine, F. E. McDonald, W. A. Neiwert and K. I. Hardcastle, J. Am. Chem. Soc., 2005, 127, 4586 CrossRef CAS.
- J.-L. Giner, J. Org. Chem., 2005, 70, 721 CrossRef CAS.
- M. A. Arnold, S. G. Duron and D. Y. Gin, J. Am. Chem. Soc., 2005, 127, 6924 CrossRef CAS.
- J. Shimokawa, T. Ishiwata, K. Shirai, H. Koshino, A. Tanatani, T. Nakata, Y. Hashimoto and K. Nagasawa, Chem. Eur. J., 2005, 11, 6878 CrossRef CAS.
- Z. D. Aron and L. E. Overman, J. Am. Chem. Soc., 2005, 127, 3380 CrossRef CAS.
- D. S. Coffey, A. I. McDonald, L. E. Overman, M. H. Rabinowitz and P. A. Renhowe, J. Am. Chem. Soc., 2000, 122, 4893 CrossRef CAS.
- L. E. Overman and Y. H. Rhee, J. Am. Chem. Soc., 2005, 127, 15652 CrossRef CAS.
- D. S. Coffey, L. E. Overman and F. Stappenbeck, J. Am. Chem. Soc., 2000, 122, 4904 CrossRef CAS.
- E. A. Colby, K. C. O'Brien and T. F. Jamison, J. Am. Chem. Soc., 2005, 127, 4297 CrossRef CAS.
- K. Takai, T. Kakiuchi, Y. Kataoka and K. Utimoto, J. Org. Chem., 1994, 59, 2668 CrossRef CAS.
- E. A. Colby and T. F. Jamison, Org. Biomol. Chem., 2005, 3, 2675 RSC.
- K. C. O'Brien, E. A. Colby and T. F. Jamison, Tetrahedron, 2005, 61, 6243 CrossRef CAS.
- M. Ishibashi, Y. Ohizumi, M. Hamashima, H. Nakamura, Y. Hirata, T. Sasaki and J. Kobayashi, J. Chem. Soc., Chem. Commun., 1987, 1127 RSC.
- W. Zhang and R. G. Carter, Org. Lett., 2005, 7, 2004.
- A. K. Mandal, J. S. Schneekloth, Jr. and C. M. Crews, Org. Lett., 2005, 7, 3645 CrossRef CAS.
- T. Andreou, A. M. Costa, L. Esteban, L. Gonzàlez, G. Mas and J. Vilarrasa, Org. Lett., 2005, 7, 4083 CrossRef.
- F. P. Liesener and M. Kalesse, Synlett, 2005, 2236 CAS.
- J. A. Marshall, G. Schaaf and A. Nolting, Org. Lett., 2005, 7, 5331 CrossRef CAS.
- M.-Y. Jang, J.-W. Kim and D.-H. Lee, Bull. Korean Chem. Soc., 2005, 26, 1497 CAS.
- B. M. Trost, J. P. N. Papillon and T. Nussbaumer, J. Am. Chem. Soc., 2005, 127, 17921 CrossRef CAS.
- (a) B. M. Trost, S. T. Wrobelski, J. D. Chisholm, P. E. Harrington and M. Jung, J. Am. Chem. Soc., 2005, 127, 13589 CrossRef CAS; (b) B. M. Trost, P. E. Harrington, J. D. Chisholm and S. T. Wrobelski, J. Am. Chem. Soc., 2005, 127, 13598 CrossRef CAS.
- H. Ishiyama, Y. Nakamura and J. Kobayashi, Tetrahedron, 2005, 62, 166.
- (a) S. P. Gunasekera, M. Gunasekera, R. E. Longley and G. K. Schulte, J. Org. Chem., 1991, 56, 1346 CrossRef CAS; (b) S. P. Gunasekera, M. Gunasekera, R. E. Longley and G. K. Schulte, J. Org. Chem., 1990, 55, 4912 CrossRef CAS.
- (a) G. R. Pettit, Z. A. Cichacz, F. Gao, M. R. Boyd and J. M. R. Schmidt, J. Chem. Soc., Chem. Commun., 1994, 1111 RSC; (b) G. R. Pettit and Z. A. Cichacz, US Pat., 1995, 5430052 Search PubMed.
- O. Loiseleur, G. Koch, J. Cercus and F. Schürch, Org. Process Res. Dev., 2005, 9, 259 Search PubMed.
- S. J. Mickel, R. Daeffler and W. Prikoszovich, Org. Process Res. Dev., 2005, 9, 259 Search PubMed.
- A. B. Smith, B. S. Freeze, M. Xian and T. Hirose, Org. Lett., 2005, 7, 1825 CrossRef CAS.
- A. B. Smith, B. S. Freeze, M. J. LaMarche, T. Hirose, I. Brouard, M. Xian, K. F. Sundermann, S. J. Shaw, M. A. Burlingame, S. B. Horwitz and D. C. Myles, Org. Lett., 2005, 7, 315 CrossRef.
- A. B. Smith, III, B. S. Freeze, M. J. LaMarche, T. Hirose, I. Brouard, P. V. Rucker, M. Xian, K. F. Sundermann, S. J. Shaw, M. A. Burlingame, S. B. Horwitz and D. C. Myles, Org. Lett., 2005, 7, 311 CrossRef.
- A. B. Smith and M. Xian, Org. Lett., 2005, 7, 5229 CrossRef.
- S. J. Shaw, K. F. Sundermann, M. A. Burlingame, D. C. Myles, B. S. Freeze, M. Xian, I. Brouard and A. B. Smith, III, J. Am. Chem. Soc., 2005, 127, 6532 CrossRef CAS.
- A. B. Smith, P. V. Rucker, I. Brouard, B. S. Freeze, S. Xia and S. B. Horowitz, Org. Lett., 2005, 7, 5199 CrossRef CAS.
- (a) I. Paterson and I. Lyothier, J. Org. Chem., 2005, 70, 5494 CrossRef CAS; (b) I. Paterson, O. Delgado, G. J. Florence, I. Lyothier, M. O'Brien, J. P. Scott and N. Sereinig, J. Org. Chem., 2005, 70, 150 CrossRef CAS.
- (a) Y. Shin, J.-H. Fournier, R. Balachandran, C. Madiraju, B. S. Raccor, G. Zhu, M. C. Edler, E. Hamel, B. W. Day and D. P. Curran, Org. Lett., 2005, 7, 2873 CrossRef CAS; (b) C. O. Kangani, A. M. Brueckner and D. P. Curran, Org. Lett., 2005, 7, 379 CrossRef CAS.
- C. Madiraju, M. C. Edler, E. Hamel, B. S. Raccor, R. Van Balachandran, G. Zhu, K. A. Giuliano, A. Vogt, Y. Shin, J.-H. Fournier, Y. Fukui, A. M. Brueckner, D. P. Curran and B. W. Day, Biochemistry, 2005, 44, 15053 CrossRef CAS.
- J. Shin and W. Fenical, J. Org. Chem., 1991, 56, 3392 CrossRef CAS.
- A. D. Rodriguez, E. Gonzalez and C. Gonzalez, J. Nat. Prod., 1995, 58, 226 CrossRef CAS.
- J. S. Kingsbury and E. J. Corey, J. Am. Chem. Soc., 2006, 127, 13813.
- K. L. Rinehart, J. Kobayashi, G. C. Harbour, J. Gilmore, M. Mascal, T. G. Holt, L. S. Shield and F. Lafargue, J. Am. Chem. Soc., 1987, 109, 3378 CrossRef.
- G. R. Pettit, J. P. Xu, J. C. Chapuis, R. K. Pettit, L. P. Tackett, D. L. Doubek, J. N. A. Hooper and J. M. Schmidt, J. Med. Chem., 2004, 47, 1149 CrossRef CAS.
- R. H. Cichewicz, F. A. Valeriote and P. Crews, Org. Lett., 2004, 6, 1951 CrossRef CAS.
- X. Jiang, J. Garcia-Fortanet and J. K. De Brabander, J. Am. Chem. Soc., 2005, 127, 11254 CrossRef CAS.
- B. S. Davidson, Tetrahedron Lett., 1992, 33, 3721 CrossRef CAS.
- P. Magnus and K. S. Matthews, J. Am. Chem. Soc., 2006, 127, 12476.
- J. W. Lane, Y. Chen and R. M. Williams, J. Am. Chem. Soc., 2006, 127, 12684.
- W. Jin and R. M. Williams, Tetrahedron Lett., 2003, 44, 4635 CrossRef CAS.
- C. Chan, R. Heid, S. Zheng, J. Guo, B. Zhou, T. Furuuchi and S. J. Danishefsky, J. Am. Chem. Soc., 2005, 127, 4596 CrossRef CAS.
- P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2005, 127, 15394 CrossRef CAS.
- P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2004, 126, 7450 CrossRef CAS.
- H. Kang and W. Fenical, J. Org. Chem., 1997, 62, 3254 CrossRef CAS.
- A. Hamasaki, J. M. Zimpleman, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2005, 127, 10767 CrossRef CAS.
- K. Warabi, S. Matsunaga, R. W. M. van Soest and N. Fusetani, J. Org. Chem., 2003, 68, 2765 CrossRef CAS.
- A. Fürstner, M. M. Domostoj and B. Scheiper, J. Am. Chem. Soc., 2005, 127, 11620 CrossRef.
- MarinLit Database, Department of Chemistry, University of Canterbury, Christchurch, New Zealand, http://www.chem.canterbury.ac.nz/marinlit/marinlit.shtml Search PubMed.
- H. Lee, H. Kim, T. Yoon, B. Kim, S. Kim, H.-D. Kim and D. Kim, J. Org. Chem., 2005, 70, 8723 CrossRef CAS.
- S. Lopez, F. Fernandez-Trillo, P. Midon, L. Castedo and C. Saa, J. Org. Chem., 2005, 70, 6346 CrossRef CAS.
- P. Basabe, S. Delgado, I. S. Marcos, D. Diez, A. Diego, M. De Roman and J. G. Urones, J. Org. Chem., 2005, 70, 9480 CrossRef CAS.
- J. Clayden, F. E. Knowles and I. R. Baldwin, J. Am. Chem. Soc., 2005, 127, 2412 CrossRef CAS.
- G. Liang, A. K. Miller and D. Trauner, Org. Lett., 2005, 7, 819 CrossRef CAS.
- S. N. Georgiades and J. Clardy, Org. Lett., 2005, 7, 4091 CrossRef CAS.
- P. Bhaket, K. Morris, C. S. Stauffer and A. Datta, Org. Lett., 2005, 7, 875 CrossRef CAS.
- N. Sudhakar, A. R. Kumar, A. Prabhakar, B. Jagadeesh and B. V. Rao, Tetrahedron Lett., 2005, 46, 325 CrossRef CAS.
- B. Zou, K. Long and D. Ma, Org. Lett., 2005, 7, 4237 CrossRef CAS.
- D. W. Jeffery, M. V. Perkins and J. M. White, Org. Lett., 2005, 7, 1581 CrossRef CAS.
- T. Ichige, S. Kamimura, K. Mayumi, Y. Sakamoto, S. Terashita, E. Ohteki, N. Kanoh and M. Nakata, Tetrahedron Lett., 2005, 46, 1263 CrossRef CAS.
- L. Yang, D. E. Williams, A. Mui, C. Ong, G. Krystal, R. van Soest and R. J. Anderson, Org. Lett., 2005, 7, 1073 CrossRef CAS.
- G. Mehta and S. Chandra Pan, Tetrahedron Lett., 2005, 46, 5219–5223 CrossRef CAS.
- T. Takanami, H. Tokoro, D. Kato, S. Nishiyama and T. Sugai, Tetrahedron Lett., 2005, 46, 3291 CrossRef CAS.
- I. R. Davies, M. Cheeseman, D. G. Niyadurupola and S. D. Bull, Tetrahedron Lett., 2005, 46, 5547 CrossRef CAS.
- A. Napolitano, I. Bruno, R. Riccio and L. Gomez-Paloma, Tetrahedron, 2005, 61, 6808 CrossRef CAS.
- E. J. Alvarez-Manzaneda, R. Chahboun, I. Barranco, E. Cabrera Torres, E. Alvareza and R. Alvarez-Manzaneda, Tetrahedron Lett., 2005, 46, 5321 CrossRef CAS.
- V. Kulcitki, N. Ungur, M. Gavagnin, M. Carbone and G. Cimino, Eur. J. Org. Chem., 2005, 1816 CrossRef CAS.
- H. Miyaoka, Y. Hara, I. Shinohara, T. Kurokawa and Y. Yamada, Tetrahedron Lett., 2005, 46, 7945 CrossRef CAS.
- T. Ogamino and S. Nishiyama, Tetrahedron Lett., 2005, 46, 1083 CrossRef CAS.
- I. Paterson, G. J. Florence, A. C. Heimann and A. C. Mackay, Angew. Chem., Int. Ed., 2005, 44, 1130 CrossRef CAS.
- S. Yu, X. Pan, X. Lin and D. Ma, Angew. Chem., Int. Ed., 2005, 44, 135 CrossRef CAS.
- K. C. Nicolaou, D. W. Kim, D. Schlawe, D. E. Lizos, R. G. de Noronha and D. A. Longbottom, Angew. Chem., Int. Ed., 2005, 44, 4925 CrossRef CAS.
- T. Mulhaupt, H. Kaspar, S. Otto, M. Reichert, G. Bringmann and T. Lindel, Eur. J. Org. Chem., 2005, 334 CrossRef.
- M. Kolympadi, M. Liapis and V. Ragoussis, Tetrahedron, 2005, 61, 2003 CrossRef CAS.
- T. Kouko, K. Matsumura and T. Kawasaki, Tetrahedron, 2005, 61, 2309 CrossRef CAS.
- E. A. Couladouros, E. N. Pitsinos, V. I. Moutsos and G. Sarakinos, Chem. Eur. J., 2005, 11, 406 CrossRef.
- X. Gao and D. G. Hall, J. Am. Chem. Soc., 2005, 127, 1628 CrossRef CAS.
- J. Justicia, J. L. Oller-Lopez, A. G. Campana, J. E. Oltra, J. M. Cuerva, E. Bunuel and D. J. Cardenas, J. Am. Chem. Soc., 2005, 127, 14911 CrossRef CAS.
- P. Wipf and T. D. Hopkins, Chem. Commun., 2005, 3421 RSC.
- M. W. B. Pfeiffer and A. J. Phillips, J. Am. Chem. Soc., 2005, 127, 5334 CrossRef CAS.
- M. Harmata and X. Hong, Org. Lett., 2005, 7, 3581 CrossRef CAS.
- J. C. Lanter, H. Chen, X. Zhang and Z. Sui, Org. Lett., 2005, 7, 5905 CrossRef CAS.
- G. Liang, A. K. Miller and D. Trauner, Org. Lett., 2005, 7, 819 CrossRef CAS.
- A. Minatti and K. H. Dötz, J. Org. Chem., 2005, 70, 3745 CrossRef CAS.
- T. J. Donohoe, H. O. Sintim, L. Sisangia, K. W. Ace, P. M. Guyo, A. Cowley and J. D. Harling, Chem. Eur. J., 2005, 11, 4227 CrossRef CAS.
- A. Le Flohic, C. Meyer and J. Cossy, Org. Lett., 2005, 7, 339 CrossRef CAS.
- F. Ding and M. P. Jennings, Org. Lett., 2005, 7, 2321 CrossRef CAS.
- F. A. Davis and J. Deng, Org. Lett., 2005, 7, 621 CrossRef CAS.
- N. Sayyadi, D. Skropeta and K. A. Jolliffe, Org. Lett., 2005, 7, 5497 CrossRef CAS.
- C. A. Mast, S. Eissler, A. Stoncius, H.-G. Stammler, B. Neumann and N. Sewald, Chem. Eur. J., 2005, 11, 4667 CrossRef CAS.
- I. Shiina, M. Hashizume, Y. Yamai, H. Oshiumi, T. Shimazaki, Y. Takasuna and R. Ibuka, Chem. Eur. J., 2005, 11, 6601 CrossRef CAS.
- X. Moreau, B. Bazan-Tejeda and J.-M. Campagne, J. Am. Chem. Soc., 2005, 127, 7288 CrossRef CAS.
- K. C. Nicolaou, D. W. Kim, D. Schlawe, D. E. Lizos, R. G. de Noronha and D. A. Longbottom, Angew. Chem., Int. Ed., 2005, 44, 4925 CrossRef CAS.
- F. Sarabia and A. Sanchez-Ruiz, J. Org. Chem., 2005, 70, 9514 CrossRef CAS.
- M. C. Carreño, R. Des Mazery, A. Urbano, F. Colobert and G. Solladié, Org. Lett., 2005, 7, 2039 CrossRef CAS.
- D. E. N. Jacquot, M. Zöllinger and T. Lindel, Angew. Chem., Int. Ed., 2005, 44, 2295 CrossRef CAS.
- J. R. Fuchs and R. L. Funk, Org. Lett., 2005, 7, 677 CrossRef CAS.
- Q. Chen, H. Deng, J. Zhao, Y. Lu, M. He and H. Zhai, Tetrahedron, 2005, 61, 8390 CrossRef CAS.
- J. E. D. Kirkham, T. D. L. Courtney, V. Lee and J. E. Baldwin, Tetrahedron, 2005, 61, 7219 CrossRef CAS.
- Y.-J. Jian, Y. Wu, L. Lib and J. Lua, Tetrahedron: Asymmetry, 2005, 16, 2649 CrossRef CAS.
- B. M. Trost, C. Pissot-Soldermann and I. Chen, Chem. Eur. J., 2005, 12, 951 CrossRef.
- N. Kotoku, H. Tsujita, A. Hiramatsu, C. Mori, N. Koizumi and M. Kobayashi, Tetrahedron, 2005, 61, 7211 CrossRef CAS.
- (a) D. Castoldi, L. Caggiano, L. Panigada, O. Sharon, A. M. Costa and C. Gennari, Angew. Chem., Int. Ed., 2005, 44, 588 CrossRef CAS; (b) D. Castoldi, L. Caggiano, L. Panigada, O. Sharon, A. M. Costa and C. Gennari, Chem. Eur. J., 2005, 12, 51.
- H. M. L. Davies and A. M. Walji, Angew. Chem., Int. Ed., 2005, 44, 1733 CrossRef CAS.
- J. T. Gupton, R. B. Miller, K. E. Krumpe, S. C. Clough, E. J. Banne, R. P. F. Kanters, K. X. Du, K. M. Keertikar, N. E. Lauerman, J. M. Solano, B. R. Adams, D. W. Callahan, B. A. Little, A. B. Scharfa and J. A. Sikorski, Tetrahedron, 2005, 61, 1845 CrossRef CAS.
- J. Uenishi and M. Ohmi, Angew. Chem., Int. Ed., 2005, 44, 2756 CrossRef CAS.
- L. K. Shubina, S. N. Fedorov, O. S. Radchenko, N. N. Balaneva, S. A. Kolesnikova, P. S. Dmitrenok, A. Bode, Z. Dong and V. A. Stonik, Tetrahedron Lett., 2005, 46, 559 CrossRef CAS.
- S. K. Jackson, S. C. Banfield and M. A. Kerr, Org. Lett., 2005, 7, 1215 CrossRef CAS.
- R. B. Andrade and S. F. Martin, Org. Lett., 2005, 7, 5733 CrossRef CAS.
- D. C. Harrowven, D. D. Pascoe, D. Demurtas and H. O. Bourne, Angew. Chem., Int. Ed., 2005, 44, 1221 CrossRef CAS.
- F. Ding and M. P. Jennings, Org. Lett., 2005, 7, 2321 CrossRef CAS.
- Y. Morita, H. Tokuyama and T. Fukuyama, Org. Lett., 2005, 7, 4337 CrossRef CAS.
- J.-F. Poisson, A. Orellana and A. E. Greene, J. Org. Chem., 2005, 70, 10860 CrossRef CAS.
- A. Srikrishna and G. Satyanarayana, Tetrahedron, 2005, 61, 8855 CrossRef CAS.
- B. M. Chanda and R. S. Sulake, Tetrahedron Lett., 2005, 46, 6461 CrossRef CAS.
- G. Papeo, H. Posteri, D. Borghi and M. Varasi, Org. Lett., 2005, 7, 5641 CrossRef CAS.
- N. K. Garg and B. M. Stoltz, Tetrahedron Lett., 2005, 46, 2423 CrossRef CAS.
- K. Fujiwara, S. Yoshimoto, A. Takizawa, S. Souma, H. Mishima, A. Murai, H. Kawai and T. Suzuki, Tetrahedron Lett., 2005, 46, 6819 CrossRef CAS.
- S. Baek, H. Jo, H. Kim, H. Kim, S. Kim and D. Kim, Org. Lett., 2005, 7, 75 CrossRef CAS.
- H. Abe, S. Aoyagi and C. Kibayashi, J. Am. Chem. Soc., 2005, 127, 1473 CrossRef CAS.
- M. Jin and R. E. Taylor, Org. Lett., 2005, 7, 1303 CrossRef CAS.
- D. Pla, A. Marchal, C. A. Olsen, F. Albericio and M. Álvarez, J. Org. Chem., 2005, 70, 8231 CrossRef CAS.
- Q. Su and J. S. Panek, Angew. Chem., Int. Ed., 2005, 44, 1223 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |