Bisintercalator natural products with potential therapeutic applications: isolation, structure determination, synthetic and biological studies
Simon
Dawson
,
John P.
Malkinson
,
David
Paumier
and
Mark
Searcey
*
Department of Pharmaceutical and Biological Chemistry, School of Pharmacy, University of London, 29–39 Brunswick Square, London, WC1N 1AX, UK. and School of Chemical Sciences and Pharmacy, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: m.searcey@uea.ac.uk; Fax: +44 1603 592003; Tel: +44 1603 592026
First published on 13th November 2006
Abstract
Covering: up to 2005
Echinomycin is the prototypical bisintercalator, a molecule that binds to DNA by inserting two planar chromophores between the base-pairs of duplex DNA, placing its cyclic depsipeptide backbone in the minor groove. As such, it has been the focus of an extensive number of investigations into its biological activity, nucleic acid binding and, to some extent, its structure–activity relationships. However, echinomycin is also the parent member of an extended family of natural products that interact with DNA by a similar mechanism of bisintercalation. The structural variety in these compounds leads to changes in sequence selectivity and biological activity, particularly as anti-tumour and anti-viral agents. One of the more recently identified marine natural products that is moving close to clinical development is thiocoraline, and it therefore seems timely to review the various bisintercalator natural products.
Simon Dawson received his MPharm degree at the University of Bath in 2004, and registered as a pharmacist in 2005. He is currently studying for a PhD at the School of Pharmacy, under the supervision of Dr Malkinson. |
Dr Malkinson was appointed as Lecturer in Pharmaceutical Science Applied to Practice at The School of Pharmacy, University of London in June 2003. He completed his BPharm at The School of Pharmacy in 1996, subsequently training and registering as a pharmacist in 1997. He returned to The School of Pharmacy and completed his PhD on the solid-phase synthesis of anti-tumour somatostatin analogues, under the supervision of Professors Istvan Toth and A. T. Florence. From 2000, he held the C. W. Maplethorpe post-doctoral teaching fellowship at the University of London, working on the synthesis and delivery of glycopeptides and glycolipid-modified bioactive peptide drugs. Current research focuses on solid-phase synthetic methodology and the synthesis of peptide conjugates and natural products with anti-tumour and anti-bacterial activity. |
David Paumier received his DEA in Chemistry in 2000 from the University of Paris V, Rene Descartes, where he studied the synthesis of amino-cyclitols as potential glycosidase inhibitors for his final year project, under the supervision of Professor Y. le Merrer. He obtained his PhD in 2004 from the University of Exeter for his work towards the total synthesis of ficellomycin under the supervision of Professor Mike Shipman. In 2005 he joined Dr Searcey's group as a post-doctoral research assistant and is currently working on the development of novel anti-cancer agents both by solution- and solid-phase synthesis. |
Dr Mark Searcey is Reader in Medicinal Chemistry in the School of Chemical Sciences and Pharmacy, University of East Anglia. He gained his first degree in chemistry at Loughborough University, and then carried out a PhD with J. B. Lee at Hatfield Polytechnic. He spent two years as a post-doctoral research assistant with the Gallacher/Brocklehurst groups at Queen Mary College, University of London, these groups being the first to describe polyclonal catalytic antibodies. In 1991, he moved to Dublin as a research scientist in St Luke's Institute of Cancer Research, studying drug–DNA interactions with Larry Wakelin. Following this, he became a research assistant and then Assistant Professor with Dale Boger at Scripps Research Institute in California. From 1999 to 2006, he was Senior Lecturer at the School of Pharmacy, University of London. His research interests are in the design and synthesis of anti-tumour agents. |
 From left to right: John Malkinson, Simon Dawson, Mark Searcey and David Paumier. |
1 Introduction
This report describes the family of natural products that bind to duplex DNA through a process known as bisintercalation. This involves the insertion of two planar chromophores between the bases, placing the group linking the chromophores in one of the grooves of the duplex. In the natural products, the linker group may be a cyclic octadepsipeptide, a cyclic thiodepsipeptide, a cyclic decadepsipeptide or a cyclic decapeptide. In all compounds studied to date, the linker is placed in the minor groove and the sequence reading abilities vary from the relatively neutral binding of the luzopeptins to the 5′-CG selectivity of echinomycin (1). Interest in these compounds was maintained for many years by the potent anti-tumour activity of echinomycin and its progression into clinical trials. However, it has also been continued by the desire to understand how these compounds target DNA and how their high affinity can be translated into biological utility. In more recent times, the anti-HIV potential of the luzopeptins 6–8 and quinoxapeptins 9–11 has stimulated interest, whereas the recent isolation of thiocoraline4 and its development by the company PharmaMar may mean that a second compound from this family progresses into clinical trial. The literature on this family of compounds is extremely large, and herein we have selectively reviewed the various family members to give an insight into their isolation, structure, synthesis, biosynthesis and biological activity.
2 Echinomycin
Isolation and structure elucidation of echinomycinThe isolation of echinomycin1 as a lipophilic antibiotic from Streptomyces echinatus sp. 1 was disclosed in 1957.1 Quinomycin A, isolated in 1961, was found to be identical to echinomycin.2 The structure of echinomycin was originally suggested to contain a 2-quinoxalinecarbonyl group on the basis of its UV absorption spectrum and to possess a peptide structure.1 It was also found to contain 2 N-CH3 and 2 C-CH3 groups. In 1959, a structure for echinomycin was proposed incorporating a cyclic depsipeptide containing L-alanine, N-methyl-L-valine, N-methyl-L-cysteine and N-methyl-D-serine. The side-chain of the N-methyl-D-serine residue forms an ester with valine, while its α-amino-group forms an amide linkage to the 2-quinoxalinecarbonyl chromophore .3 The symmetrical nature of the molecule was completed by an unusual dithiane bridge between the cysteines.3 This structure was predicted based on the elemental analysis. The development of NMR as a tool for structure elucidation meant that it later became apparent that echinomycin was not symmetrical, and the dithiane bridge was revised to an equally unusual dithioacetal .4
Synthesis and biosynthesis of echinomycin
To date, there are no descriptions of a chemical synthesis of echinomycin. Presumably, the simpler chemistry involved in the development of routes to triostin A, which contains a disulfide bridge rather than the more synthetically challenging thioacetal, along with the ready commercial availability of the natural product from fermentation, has led to a lack of interest. Synthesis has been limited to modifications of the natural product structure. For example, recently, attempts to increase the anti-tumour activity of echinomycin focused on the formation of a methyl sulfonium analogue 13 that showed enhanced potency in tumour cell lines compared with the parent compound.5Sulfone and sulfoxide analogues were significantly less active. The same authors have also looked at an echinomycin analogue in which the dithioacetal bridge was replaced by a methylene dithioether (although this is strictly an analogue of triostin A).6 The resulting compound 14 has a similar activity to the natural product and, as noted for the thioacetal bridge, oxidation to the sulfoxide and sulfone led to a precipitous loss of activity.6
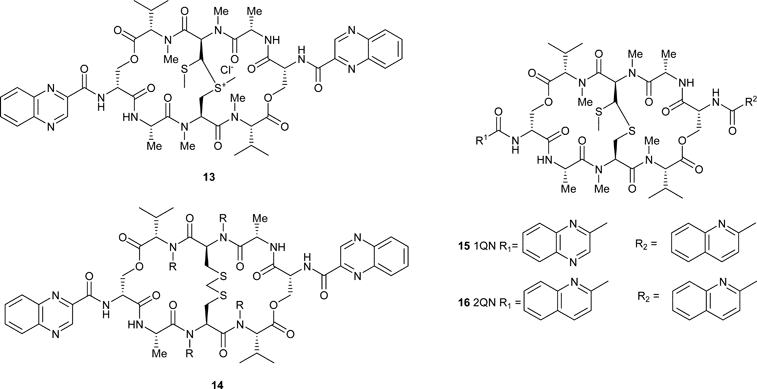
The biosynthesis of echinomycin has been studied in somewhat greater detail. In 1969, the first analogue of echinomycin (quinazomycin) was described, produced by feeding of quinazol-4-on-3-acetic acid, such that one of the natural quinoxaline chromophores was replaced.7 Later work described a cell-free system to produce an analogue in which both of the chromophores were replaced, and this compound was given the name biquinazomycin.8 This approach to the generation of analogues was also adopted somewhat later by Waring and co-workers to study structure–activity relationships of the natural products.9 Thus analogues of echinomycin were produced in which one or both of the chromophores were replaced by a quinoline moiety (see compounds 15 and 16).10 These compounds were studied for their ability to bind to DNA.11 Interestingly, the compound in which both chromophores were replaced by their quinoline isosteres showed a lack of sequence selectivity that was not noted for the compound containing one quinoline and one quinoxaline, which mirrored echinomycin more closely. This decrease in sequence selectivity has been noted for sandramycin and the luzopeptins, which carry substituted quinoline chromophores.12 It has also been suggested that triostin A2 is a natural precursor to echinomycin, as Streptomyces echinatus is able to convert the triostins into the quinomycin antibiotics.13
Feeding studies utilising 15N-labelled amino acids combined with NMR, demonstrated that the N1 and N4 of the quinoxaline chromophores were derived from the indole and amino groups of tryptophan.14 More recently, studies have focused on isolating the non-ribosomal peptide synthetases involved in the synthesis of both echinomycin and triostin A. These have demonstrated for the first time a functional interaction between non-ribosomal peptide synthesis and fatty acid biosynthesis that leads to incorporation of the quinoxalinechromophore .15
Biological activity of echinomycin
Echinomycin was found to be a potent anti-bacterial agent against Gram-negative organisms and to have good anti-tumour activity.1 In 1974, it was demonstrated by viscometric techniques that echinomycin binds to duplex DNA by bisintercalation.16 The model of binding, with the compound inserting the quinoxaline chromophores between the bases of DNA and forming a two-base-pair sandwich, with the cyclic depsipeptide positioned in the minor groove, was supported by the subsequent structural studies by Rich17 and Feigon.18 This novel mode of action, along with the potent anti-tumour activity of the natural product, has stimulated extensive studies of the DNA binding and biological activity of echinomycin. A full and comprehensive review of these studies is outside the scope of the current report. Readers are directed to an earlier review of bisintercalators for an overview of the field to 1986.19 Bailly and Waring have reviewed the study of echinomycin and other duplex-binding compounds using modified DNA substrates to further understand the contribution of base structure to sequence selectivity and binding.20
Echinomycin was originally shown to exert its biological effects as a consequence of inhibition of RNA polymerase.21 The subsequent discovery of bisintercalation suggests that the echinomycin–DNA complex can form a block to progression of the enzyme, acting as an inhibitor of transcription. Studies of synthetic polynucleotides showed that the natural product binds well to polydG·polydC and poly(dGdC), but poorly to poly(dAdT), with little binding to polydA·polydT and polydI·polydC.22
This sequence selectivity has been confirmed through the use of footprinting, possibly the most powerful technique for the study of ligand–DNA interactions, and one that has been developed extensively in the studies of bisintercalator natural products. Footprinting involves the cleavage of radiolabelled DNA with either an enzymic (usually DNase I),23 photolytic24 or chemical25nuclease (usually MPE-Fe(II), although the current authors developed an alternative chemical nuclease called FTP126). The DNA is cut alone or in the presence of increasing concentrations of ligand, attempting to maintain “single hit kinetics”, i.e. that each DNA strand is cut once. The cleaved DNA is then subjected to electrophoretic separation on a polyacrylamide gel. In the presence of a DNA-binding ligand, gaps in the resulting ladders reveal the sequences that are not cleaved by nuclease, i.e. the sites at which the drug is bound. Drew and co-workers described the application of this technique to echinomycin in 1984, using the nuclease DNase I, and showed that footprints appeared in the tyrT fragment at sequences containing a 5′-CG.23 In the same year, van Dyke and Dervan described the use of the chemical nuclease MPE-Fe(II) (methidium propyl-EDTA-Fe(II)), which allows a precise analysis of binding sites that were four base-pairs in length, each with a central 5′-CG.25 The preferred high affinity flanking base-pairs were A and T, such that the best binding sites were 5′-ACGT and 5′-TCGT. Developments in footprinting have meant that studies of echinomycin binding to nucleosome core particles27 and also the kinetics of binding to various sequences28 have been disclosed. These latter studies are particularly interesting, as they allow the movement away from the surfactant-sequestration technique (which has been extremely valuable in the study of kinetics of binding to natural and synthetic DNA29) into a more defined analysis of the relationship of sequence context to DNA binding kinetics. Investigations of the kinetics of binding are valuable, as they may be related to the ability of the molecule to inhibit RNA polymerase.
The first structural analyses of echinomycin and triostin A binding to 5′-CGTACG demonstrated only small differences in the complexes of the two natural products with this sequence, notably due to the shorter cross-bridge of echinomycin.17 Both compounds had a hydrogen bond between alanine and guanine and induced Hoogsteen base-pairing in the central thymine and adenine bases. Solution footprinting studies using diethyl pyrocarbonate to highlight hyper-reactive sites in the DNA upon binding were suggested to support the formation of Hoogsteen base-pairs on echinomycin binding.30 The effect of this reagent was later suggested to be due to unwinding of the helix rather than Hoogsteen base-pair formation.31 In a series of experiments, Waring and co-workers showed that Hoogsteen base-pairing was not necessary for echinomycin binding, through the use of DNA containing base analogues (2′-deoxy-7-deazaadenosine and 2′-deoxy-7-deazaguanosine) that cannot form Hoogsteen base-pairs.32 In several NMR-based studies, Feigon and co-workers showed that while Hoogsteen base-pairing may occur, it tended to be at the ends of the oligonucleotides studied and internal base-pairs were dynamic, alternating between open, Watson–Crick or Hoogsteen forms.33 More recent crystal structures have again demonstrated the formation of Hoogsteen base-pairs.34 Feigon's NMR studies also demonstrated that differences in sequence can lead to variations in cooperativity of binding. Echinomycin bound to the sequence 5′-[d(ACGTACGT)2] in a cooperative fashion, with two molecules present even at low concentrations, whereas the binding to 5′-[d(TCGATCGA)2] was non-cooperative.35 Examining the exchange of imino protons in the echinomycin–DNA complex demonstrated that the base-pairs within the intercalation site are held in a “vice-like” grip by the natural product.36
An in-depth study of the thermodynamic profile of echinomycin binding to DNA has been carried out using differential scanning calorimetry and UV thermal denaturation experiments.37 The outcome of the work was to suggest that the binding of echinomycin to DNA is essentially entropy-driven and that, enthalpically, hydrogen bonding and van der Waals interactions also make an important contribution.
As noted above, echinomycin is a potent anti-tumour agent and is a natural product that has been entered into clinical trials. Phase I trials established a maximum tolerated dose for echinomycin of 1.8 mg m–2, with toxicities of nausea, vomiting, reversible liver abnormalities and allergic reactions.38 Phase II trials were uniformly disappointing, with minimal to no activity against cervical squamous cell carcinoma,39 advanced ovarian tumours,40 soft tissue sarcomas,41 advanced breast cancer42 and central nervous system malignancies.43
3 The triostins
Isolation and structure elucidation of the triostinsThe triostins were isolated from a relative of Streptomyces aureus designated S-2-210.44,45 Early chromatographic45 and degradative46 studies showed that there were three similar quinoxaline components in the isolated mixture, and these were named triostins A (2), B (17) and C (18). They were, however, not present in equal amounts; the majority of the mixture consisted of triostin C.45 Degradation by acid hydrolysis supported the theory that these antibiotics were structurally related, as had been implied by the results of infrared spectroscopy.45,46 This method showed triostin C to consist of quinoxaline-2-carboxylic acid, D-serine, L-alanine, N-methyl-L-cysteine and N,β-dimethyl-L-leucine residues.46 Later NMR studies refined the configuration of this last amino acid as N,γ-dimethyl-L-alloisoleucine ((2S,3R)-3,4-dimethyl-2-methylaminopentanoic acid), and that it was linked to the hydroxyl group of D-serine.47,48 Thus, a cyclic octadepsipeptide structure with a disulfide cross-link was proposed.

Triostin A (2) differs from this structure by one amino acid; N-methyl-L-valine instead of N,γ-dimethyl-L-alloisoleucine.46 Confirmation of this structure by NMR was disclosed in 1975, demonstrating the presence of the disulfide cross-link that had been theorised for triostin C49 and which sets the triostins apart from the quinomycins (e.g.echinomycin, see above), which utilise a thioacetal instead of a disulfide.4
Apart from the isolation45 and structure elucidation by acid hydrolysis,46 very little work on triostin B (17) has been disclosed. In structure, this compound was reported to differ from triostin A only by one amino acid residue; N-methyl-L-isoleucine instead of N-methyl-L-valine.46 This was later revised by the same authors to N-methyl-L-alloisoleucine.50
Synthesis and biosynthesis of the triostins
Soon after the isolation of echinomycin and the triostins, a number of groups investigated the synthesis and biological activity of simple quinoxaline analogues with limited success.51–53 In order to investigate structure–activity relationships in the triostins, Olsen and co-workers began an investigation into synthetic approaches to the natural product by focusing on TANDEM, the N-demethylated analogue 3 (Triostin A N-DEMethylated).54 The initial synthesis involved a linear route to a tetradepsipeptide precursor 19 in which Boc-Cys(Acm)-Val-OH† was coupled to Cbz-D-Ser-Ala-OTce through an ester bond to the side-chain of serine. In a convergent approach, this tetrapeptide precursor was subsequently deprotected at either the cysteine α-amino group or the alanine carboxyl group, then dimerised to give the linear octadepsipeptide 20 incorporating both ester bonds (Scheme 1). Cyclisation by treatment with N-hydroxysuccinimide/dicyclohexylcarbodiimide (NHS/DCC) under basic conditions was followed by Acm-deprotection and oxidation with iodine in methanol to form the disulfide bridge. Further deprotection and acylation with 2-quinoxaloyl chloride gave TANDEM.55 This methodology was also used to produce a bis-L-serine analogue of TANDEM, which showed no appreciable binding to DNA.56 A subsequent investigation of a different route to TANDEM, which sought to avoid problems of racemisation on formation of the linear depsipeptide , showed little improvement in either yield or avoidance of racemisation.57
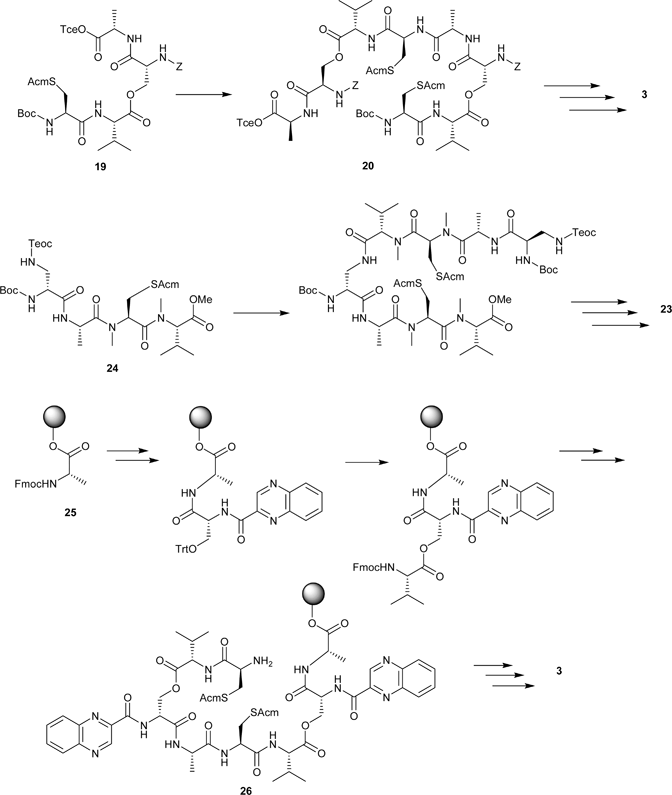 | ||
| Scheme 1 Top: Olsen’s synthesis of TANDEM.55 Middle: Boger’s synthesis of azatriostin A.64 Bottom: The Searcey/Malkinson solid-phase synthesis of TANDEM.65 | ||
The synthesis of triostin A by Chakravarty and Olsen used a similar strategy to the above, but with significant modification to the method, due to the incorporation of N-methylamino acids.58 Difficulties related to this were encountered with the Tce ester of the linear octadepsipeptide, which could only be partially deprotected, with fragmentation under more vigorous conditions. By cleaving the Tce ester earlier in the synthesis (at the tetradepsipeptide stage) and fragment coupling as in the TANDEM synthesis, the octadepsipeptide with a free carboxyl group was obtained. Cyclisation with NHS/EDC, subsequent disulfide formation, and introduction of the chromophores using quinoxaloyl chloride gave the natural product.58
Syntheses by Shin and co-workers of triostin A, TANDEM, and the S,S′-dibenzyl analogue 21 lacking the disulfide linkage,59–61 involved protection of the C-terminal D-serine as the Tce ester, which in the original synthesis of triostin A had been difficult to remove from alanine at the octadepsipeptide stage.58 However, in this case no such difficulties were encountered.59
In order to investigate further the role of the alanine residues in the binding of TANDEM to DNA, an analogue 22 was synthesised which contained L-lactic acid rather than alanine.62 Ester bond formation between Cbz-D-Ser-OH and L-lactic acid p-bromophenacyl ester was achieved using established methodology,63 and synthesis continued to the final product 22 in the usual manner.56
Azatriostin A 23 is an analogue of triostin A in which the D-serine residues are replaced by 1,3-diaminopropionic acid (Dpr), hence the two rotationally equivalent halves of the octapeptide ring are joined by amide linkages rather than esters.64 This compound was produced by a synthesis in which each synthetic intermediate could be purified by acid/base liquid–liquid extractions (Scheme 1), and hence lends itself to a combinatorial approach.64 Unlike previously described syntheses, the first step was a coupling of Boc-MeCys(Acm)-OH to the methyl ester of N-methylvaline, followed by deprotection of the N-terminus and coupling of Boc-Ala-OH. Orthogonally protected D-Dpr was subsequently coupled to this protected tripeptide to form the appropriate tetrapeptide precursor 24. Deprotection and fragment coupling was followed by disulfide formation and cyclisation in that order, since reversal of these steps yielded an unstable intermediate.64
An alternative approach to combinatorial synthesis of these compounds is to use a solid-phase methodology. This allows the rapid production of analogues in which each amino acid residue can be altered, or the symmetry of the natural compounds removed.65 The analogue TANDEM has been synthesised using a solid-phase approach starting from Wang resin-bound Nα-Fmoc-protected L-alanine 25 and assembling the depsipeptide in a stepwise linear fashion (Scheme 1). Each peptide coupling was carried out using HBTU and HOBt, in the presence of DIEA. Esterification was achieved using DIC and the chromophores were introduced using quinoxaline-2-carbonyl chloride. In contrast to previously described syntheses, each chromophore was attached prior to the esterification reaction, rather than after cyclisation. In the first-generation synthesis, disulfide formation was achieved whilst the linear peptide was still on the resin, which almost eliminates the risk of intermolecular disulfide formation.65 However, subsequent cyclisation of this compound after cleavage from the resin gave a low yield, possibly due to steric constraints imposed by the disulfide. In the second-generation synthesis, the linear peptide 26 was cleaved from the resin and then cyclised in solution, followed by disulfide formation. This gave a more satisfactory yield.65
The solid-phase approach has also been used to synthesise des-N-(tetramethyl)azatriostin A 27, the demethylated analogue of azatriostin.66 In addition to the original quinoxaline chromophores, this synthesis allows various nucleobase-substituted analogues to be produced, which are being evaluated in DNA binding studies.66
The strains of streptomycetes from which the triostins were originally isolated have also been used for their biosynthesis.67–69 Yoshida and Katagiri grew Streptomyces s-2-210L in a sodium nitrate–maltose medium and, by supplementing with radiolabelled amino acids, demonstrated that the residues in triostins A and C were all derived from the appropriate L-amino acid. However, the point at which L-Ser is inverted into its D-enantiomorph was not fully determined.67 In a similar manner, the source of the N-methyl groups on the valine and cysteine residues in triostin A was found to be methionine, and the quinoxalinechromophore was found to be derived from tryptophan.67–70Triostins A and C were found to be produced simultaneously by Streptomyces s-2-210L67 and Streptomyces triostinicus,69 however, unlike the original isolation study,45 the majority of the mixture was found to consist of triostin A. Novel analogues of the triostins were produced by supplementing S. triostinicus cultures with a variety of halo- or amino-substituted quinoxaline-2-carboxylic acids, which were incorporated as the chromophores.67,68 Additionally, a quinoline and a 6-fluoro-substituted quinoxaline analogue were generated by supplementing with quinoline-2-carboxylic acid and DL-5-fluorotryptophan respectively (28, 29).68–70 This last discovery prompted the suggestion that the carbon atom at position 5 of the indole ring of tryptophan ends up at position 6 of the triostin ring system.69
Biological activity of the triostins
Initial studies44,71 demonstrated that the triostins had significant anti-bacterial and anti-tumour activity, and, similar to echinomycin and the quinomycins, further work proposed that their mechanism of action was by bifunctional intercalation into DNA.72,73 Although footprinting patterns are similar to echinomycin, triostin A displays less preference for (G + C)-rich DNA species,73,74 leading to the proposal that the quinoxaline chromophores are not solely responsible for sequence specificity.73 The crystal structure solution of a DNA–triostin A complex showed that the depsipeptide ring adopts a roughly rectangular shape, with the disulfide bridge pointing away from the binding site.75 Previously, analogues of TANDEM in which the configuration of the D-Ser residue was inverted had been shown to have minimal or no binding to DNA.76 The triostin A complex showed that the orientation of the D-Ser amino group allows the chromophores to project parallel to each other at right angles from the peptide backbone, bracketing a CG base-pair in the minor groove of DNA.75 In addition, the crystal structure of triostin A bound to DNA was not symmetrical; the disulfide bond becomes skewed towards one end of the molecule, and the valine carbonyl groups point in opposite directions. The valine side-chains lie at the opposite corners of the rectangle to the quinoxaline chromophores, with the N-methyl groups pointing in towards the nucleic acid surface. Alanine (on the long sides of the rectangle) also has both its carbonyl groups and methyl side-chains pointing towards the DNA.17,77,78
Structural features that contribute to DNA binding have also been illuminated by comparison of triostin A with the synthetic, N-demethylated analogue TANDEM. While maintaining bifunctional intercalation, TANDEM binds with high affinity to AT-rich DNA.76 Later DNase I footprinting studies identified the exact binding site as TpA.79 It was suggested that the structural change induced by removal of the N-methyl groups exposes the NH-amide groups of L-Ala, and these could interact with the 2-keto groups of thymine.80 Extensive structural studies of [N-MeCys3,N-MeCys7]TANDEM (or CysMeTANDEM 30) have been carried out both in solution and on the solid phase. CysMeTANDEM has been shown to bind in a very similar manner to TANDEM, in that it bisintercalates a TpA step with approximately the same affinity.79,81 It has also been suggested that the N-methyl groups of cysteine play very little part in the binding to DNA, as they have been shown to project away from the minor groove.80–82 For example, an analogue in which cysteine residues were replaced entirely by alanine (i.e. lacking a disulfide) showed identical DNA binding specificity to TANDEM, although with much lower affinity. This implies that the role of cysteine is purely to hold the structure rigid via the disulfide bridge, and hence improve binding strength by reducing unfavourable entropic effects.6,80,82
Like triostin A in solution, CysMeTANDEM has been shown to adopt a symmetrical conformation when bound to DNA, stabilised as before by van der Waals forces , hydrogen bonding and stacking interactions.82 The Ala methyl group again appears critical to stability, forming van der Waals interactions with C1′ and C2′ of thymine and O4′ of adenine, in a manner analogous to triostin A binding.82 This time, however, the valine side-chains undergo less rotation and are able to form strong van der Waals interactions between one methyl group and O2 of thymine.82,83 Hydrogen bonds essential to the binding of CysMeTANDEM are formed between the NH groups of the alanines and N3 of the adenines.82–84 An analogue of TANDEM with lactic acid substituted for L-Ala demonstrates this point by having very little affinity for DNA, even at concentrations over double that of TANDEM, and approaching the limits of its solubility.62,84
To investigate the sequence specificity of both CysMeTANDEM and the natural product, Addess and Feigon studied the binding to DNA in which inosines were substituted for guanines.83 It was expected that the removal of guanine would eliminate binding of triostin A and favour binding of CysMeTANDEM, since the CpI step lacked exocyclic amino groups to form hydrogen bonds. This was borne out by the NMR results, which supported earlier findings that suggested both pairs of intermolecular hydrogen bonds were required for sequence-specific binding of triostin A to CpG.78,83 Binding to CpI was analogous to that with TpA, with hydrogen bonds between the NH group of Ala and N3 of inosine. In addition, the Ala methyl made van der Waals contacts with C1′ and C2′ of cytosine and O4′ of inosine.83
It appears, however, that the reason for specificity of the natural product and its synthetic analogue is reliant on more than just hydrogen bonding. A second study confirmed the importance of the guanine 2-amino group to the binding of triostin A, but also showed that replacing adenine with diaminopurine caused triostin A to bind to TpD sites with greater strength than it does to CpG.84 This is in spite of the fact that both sequences contain the same number of hydrogen bonding sites. The 2-aminopurine group was also previously thought to prevent binding of TANDEM to CpG by hindering fitting of the depsipeptide backbone into the minor groove. However, the binding of TANDEM to TpA appears to be completely unaffected by the substitution of adenine with diaminopurine.84
The sequence context of binding for CysMeTANDEM is also important. Footprinting studies have shown that 5′-ATAT is the binding site with highest affinity for this compound.85 The lowest affinity site was 5′-TTAA, while 5′-CTAG was amongst the highest affinity sites. Free energy simulations have shown that when faced with identical hydrogen bonding opportunities with the minor groove, these compounds intercalate at sites with the best stacking properties.86 For example, CysMeTANDEM should intercalate TpA and CpI with equal strength, with respect to hydrogen bonding, but unfavourable stacking interactions with bases neighbouring CpI steps lead to weaker binding. This theory would also explain why binding of triostin A to CpG is less tight than the binding of TANDEM to TpA,83 even though the former is stabilised by four hydrogen bonds.
The quinoxaline antibiotics have been shown to inhibit the action of a restriction enzyme.87 Bacteriophage ØX714 DNA is usually digested by HpaI at three sites with a recognition sequence of GTTAAC. Bisintercalation by echinomycin, triostin A, triostin C, TANDEM and other synthetic analogues was shown to inhibit cleavage at two of the three sites, the first being AT-rich and the second GC-rich. The cleavage kinetics of the third site could not be measured.87 Triostin A showed roughly equal binding to both of the sites and TANDEM showed preference for the AT-rich site, as one would expect. Interestingly, the bisquinoline analogue of triostin A showed preference for the GC-rich site over the AT-site; triostin C on the other hand bound much more weakly than triostin A, which is in contrast to previous findings.87 This suggests that restriction enzyme cleavage inhibition by triostins must also be affected by other factors apart from DNA binding strength and selectivity.
4 Thiocoraline and other cyclic octa(depsi)peptide bisintercalators
4.1 Thiocoraline
The cyclic depsipeptide antibiotic of most significant current interest for development as an anti-tumour agent of potential clinical value is thiocoraline (4). It is currently undergoing pre-clinical evaluation by PharmaMar, in an effort to optimise dosing schedule and drug delivery prior to its progression into Phase I clinical trials (from www.pharmamar.com, June 2006).
Thiocoraline is the most recently isolated and characterised of the cyclic depsipeptide natural products discussed so far, and its rapid investigation and biological evaluation are a reflection of its potential as a novel cancer drug. It was first isolated in 1997 by Romero and co-workers from strain L-13-ACM2-092 of Micromonospora marina, an actinomycete growing on soft coral in the Indian Ocean near Mozambique.88 Cytotoxicity assay-guided fractionation of the ethyl acetate extract of the mycelial cake from 4.5 L of fermentation broth, employing normal then reversed-phase chromatography and preparative TLC, yielded 40 mg of thiocoraline.
Initial NMR-based structural elucidation of thiocoraline identified it as a two-fold symmetrical bicyclic octadepsipeptide, bearing two aromatic chromophores, similar to the quinoxaline antibiotics described above.89Thiocoraline differs from the depsipeptide antibiotics discussed above in that it is a thiodepsipeptide, i.e. it incorporates two thioester linkages in place of the ester linkages seen in e.g.triostin A. Each thioester is formed between the side-chain thiol of the chromophore -bearing cysteine residue and the carboxyl group of an unusual N,S-dimethylcysteine residue. Thiocoraline also incorporates glycine residues in place of the L-alanine residues of triostin A, as well as the same N-methylcysteine residues, through which the disulfide cross-link is formed. The 3-hydroxyquinoline-2-carbonyl chromophores are the same as seen on sandramycin. The low polarity of thiocoraline confers solubility in ethyl acetate and chloroform, with only sparing solubility in methanol and insolubility in water.89
The absolute and relative stereochemistry of thiocoraline were not defined as part of the initial structural elucidation. Triostin A and echinomycin have D-stereochemistry at the stereogenic centre of the chromophore -bearing residue (D-serine in both cases) and L-stereochemistry at the α-carbons of all the remaining residues. Boger and Ichikawa,90 in the first synthesis of thiocoraline, anticipated the same stereochemistry at each of the corresponding stereogenic centres in thiocoraline. The spectral characteristics and properties of their synthetic product were identical to those of the natural product, confirming the proposed structure and establishing its absolute and relative stereochemistry.90
Synthetic efforts to date are confined to the solution-phase studies of the Boger group.90,91 Their synthesis involved the convergent synthesis and condensation of appropriately protected tetradepsipeptide building blocks with late stage formation of the troublesome and labile thioester. Basic reaction conditions must be avoided subsequent to thioesterification due to the risk of competing cleavage and β-elimination. Assembly of the linear octadepsipeptide precursor was followed by disulfide formation and finally macrolactamisation. Final stage deprotection and chromophore introduction was conducive to the synthesis of symmetrical chromophore -substituted analogues. The synthetic thiocoraline was identical to the natural product and demonstrated highly potent (IC50 = 200 pM) cytotoxicity against a murine leukaemia cell line .
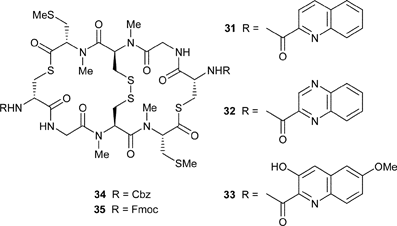
Analogues have been synthesised in which the native 3-hydroxyquinoline-2-carbonyl chromophore was replaced by either quinoline-2-carbonyl (31), quinoxaline-2-carbonyl (32; as for triostin A and echinomycin) or 3-hydroxy-6-methoxyquinoline-2-carbonyl (33; as for the luzopeptins) chromophores. Analogue 33 showed similar potent activity to thiocoraline, but 31 and 32 were less cytotoxic, although they did bind to DNA in a similar manner to thiocoraline. The bis-Cbz- and bis-Fmoc-protected precursors (34 and 35 respectively) were inactive.
Thiocoraline demonstrates potent anti-bacterial activity (MIC = 0.05 µg mL–1) against Gram-positive bacteria such as Staphylococcus aureus, but essentially no activity against Gram-negative organisms.88 It has a wide spectrum of anti-proliferative activity against human non-small-cell lung carcinoma, breast, colon, renal and melanoma cancer cell lines in vitro,88,92 as well as against human carcinoma xenografts in vivo,93 and is highly potent, with IC50 values for many cell lines ranging from high nanomolar down to high picomolar.
Thiocoraline most likely exerts its anti-proliferative activity through a variety of mechanisms, arresting colon tumour cells in the G1 phase of the cell cycle and decreasing the rate of progression from S to G2/M phases.92 It does not inhibit topoisomerase I or II, nor does it cause any DNA cleavage or alkylation, but it does cause inhibition of DNA polymerase α, possibly by the formation of a ternary enzyme–DNA–thiocoraline complex.88,92
The binding of thiocoraline to DNA has been investigated by Boger and co-workers.91 It binds to DNA with high affinity (KB = 2.6 × 106 M–1) but little or no sequence selectivity, in contrast to the quinoxaline antibiotics. This is most likely a consequence of the depsipeptide sequence rather than the chromophores. Thiocoraline unwinds duplex DNA91,92 in a manner most indicative of a bisintercalative mode of binding, similar to other related natural products in the class.
Pre-clinical evaluation of thiocoraline, aided by the development of sensitive assays for the compound,94 has shown a half-life in human plasma of around 4 h, with the majority of biotransformation to inactive metabolites occurring in the liver by the CYP3A4 cytochrome P450 isozyme , followed by phase II metabolism and clearance.95 This may present a risk of drug–drug interactions and inter-patient variability (genetic and gender-based) in its clinical use. Tumours expressing CYP3A4 may also be less susceptible to thiocoraline. The potential for thiocoraline resistance also exists as, like doxorubicin, it is a substrate for P-glycoprotein, and doxorubicin-resistant tumour cell lines expressing high levels of P-glycoprotein are also resistant to thiocoraline.92
4.2 BE-22179
The cyclic thiodepsipeptide antibiotic BE-22179 (36) bears very close structural resemblance to thiocoraline4, differing only in the substitution of the N,S-dimethylcysteine residues with unusual N-methyldehydroalanine residues. It was isolated as a part of a screening program to identify novel topoisomerase inhibitors from Streptomyces sp. A22179 (a strain resembling Streptomyces gangtokensis) found in soil from Mt Myogi in the Gumma prefecture of Japan.96 Methanolic extraction of the mycelial cake from filtration of the fermentation broth and several solvent extraction and washing steps followed by chromatography on silica gel produced material for assessment. Compound 36 has a similar solubility profile to thiocoraline. NMR-based structural elucidation was complicated by its adoption of several similar conformers in solution, but was simplified by the synthesis of a diacetate derivative.91,96
In concert with their development of a synthetic route to thiocoraline, confirmation of the structure of BE-22179 and establishment of its stereochemistry was achieved by Boger and co-workers, by comparison of the synthetic and natural products.90,91Thiocoraline was converted to BE-22179 by NaIO4-mediated oxidation followed by elimination.
BE-22179 demonstrates strong activity against Gram-positive bacteria (including S. aureus) but is inactive against Gram-negative bacteria. Initial evaluation of anti-proliferative activity demonstrated very good in vitro cytotoxic activity against P388, L1210 and MKN-45 cell lines (IC50ca. 3–6 nM) and interperitoneal (i.p.). administration of BE-22179 improved survival time nearly 3-fold over controls of mice transplanted with L1210 (murine leukaemia) cells at one quarter of the toxic dose.96
Unlike thiocoraline, BE-22179 is a highly potent inhibitor of DNA topoisomerase II (IC50 = 0.03 µM), being ca. 200-fold more active than echinomycin.96,97 It is likely that it binds to and unwinds DNA in a bisintercalative manner similar to thiocoraline.91,97 However, inhibition of the DNA-relaxing activity of topoisomerase II is independent of this conformational change, in contrast to echinomycin, for which inhibition of DNA relaxation is most likely due to bisintercalation. BE-22179 most probably interacts directly with topoisomerase II, or prevents binding of the enzyme to its DNA recognition site .97 BE-22179 is at least 100-fold less active as an inhibitor of topoisomerase I than II, and is less active than echinomycin as an inhibitor of RNA synthesis. In an analogous manner to thiocoraline, BE-22179 binds to DNA with high affinity and no real sequence selectivity; it does not alkylate, cleave or cross-link DNA, in spite of the presence of the electrophilic exo-methylene group.91
4.3 Other related anti-tumour cyclic depsipeptides
SW-163C (37) and E (38) were recently isolated from the SNA15896 strain of Streptomyces sp., an actinomycete found in a soil sample collected from Yuuki City in the Ibaraki prefecture of Japan.98 Both showed potent anti-bacterial activity against a range of predominantly Gram-positive organisms, with SW-163E showing greater activity. SW-163E also showed greater (around 100-fold) cytotoxicity than SW-163C against a variety of tumour cell lines in vitro, with IC50 values in the low nM range.
Structural elucidation of these depsipeptides highlighted several similarities to known cyclic depsipeptide antibiotics.99 They are two-fold symmetrical bicyclic octadepsipeptides and bear the same 3-hydroxyquinoline-2-carbonyl chromophores as thiocoraline and BE-22179, but with ester rather than thioester bonds as part of the depsipeptide ring. The cross-link is mediated by a disulfide in SW-163C and a thioacetal in SW-163E (similar to triostin A and echinomycin respectively). The most unusual feature of these antibiotics is the presence of two strained N-methyl-3-methylcyclopropanecarboxylic acid residues. Similar structural motifs can be found in the quinoxapeptins (vide infra). The stereochemical configurations of SW-163C and E have yet to be determined, although they are likely to bear close resemblance to other related compounds in the class.
5 The cyclic deca(depsi)peptides
5.1 Sandramycin
In 1989, sandramycin (5) was isolated from a culture broth of Nocardioides sp. (ATCC 39419) through a sequence of solvent partition and chromatographic procedures.100 It was obtained as a white solid, soluble in chloroform, dichloromethane and ethyl acetate. The molecular formula, C60H76O16N12, was established by elemental analysis and high resolution mass spectrometry.100 Further structural elucidation was performed using UV spectroscopy, NMR experiments and degradation studies.101Sandramycin proved to be a cyclic decadepsipeptide with a two-fold axis of symmetry and two 3-hydroxyquinaldic acid chromophores. 1H, 13C NMR and COSY experiments led to the identification of the amino acids present, while HMBC and COSY data revealed the nature of the heteroaromatic chromophore . The stereochemistry of each optically active amino acid was established by GC and HPLC chiral analysis of the acid hydrolysate (Sar, Gly, L-Ser, N-methyl-L-Val, L-pipecolic acid). NOE studies and HMBC experiments were used to establish the amino acid sequence. The symmetrical nature of the natural product was revealed by the molecular formula having twice the number of carbons and protons observed by NMR spectroscopy. The sites of dimerisation and attachment of the chromophores on the D-Ser residues were established using HMBC and COLOC experiments, while an IR absorption band at 1748 cm–1 showed that the two core units were linked end to end by ester bonds.101
In 1996, Boger and co-workers reported the first solution-phase total synthesis of sandramycin.102 Key strategic elements of this synthesis include a convergent assembly of the symmetrical pentadepsipetide moieties followed by coupling, macrocyclisation and a late stage introduction of the chromophore to allow straightforward access to several analogues with diverse intercalating groups. The assembly of the pentadepsipeptide was performed in a concise fashion with minor racemisation observed.102 The ester linkage was established towards the end of the synthesis to prevent hydrolysis. Attempts to couple the pentadepsipeptides and perform the subsequent ring closure in one step led to small amounts of the cyclic product along with multiple oligomers and higher order macrocycles. As a consequence, the authors opted for a two-step strategy. Thus, using EDC and HOBt in presence of NaHCO3, the linear peptide was isolated in 86% yield. Upon treatment with PPA, the linear peptide underwent cyclisation to provide the corresponding decadepsipeptide in a 90% yield. Further deprotection of the amino functionalities and subsequent coupling of the 3-hydroxyquinoline-2-carboxylic acid chromophores provided (–)-sandramycin, after cleavage of the remaining protective groups.102 The synthesis allowed the development of routes to several analogues for structure–activity relationship studies.103–105
Sandramycin was shown to exert strong anti-microbial activity against Gram-positive Bacillus subtilis, Staphylococcus aureus and Streptococcus faecalis (MIC 0.012 to 0.024 µg mL–1).100 As an anti-tumour agent, sandramycin gave a moderate response in vivo in the P388 tumour model, with values comparable to those observed for luzopeptin A.100Sandramycin also inhibits HIV-1 reverse transcriptase (IC50 130 nM).103 In many respects, sandramycin mirrors luzopeptin A (6) in its mode of interaction with DNA.102 Both compounds target the minor groove of DNA and bind at least 10 times more tightly than echinomycin.106 They have also proven to exert comparable in vitro cytotoxicity (IC50 6 to 0.02 nM). Binding studies revealed that sandramycin possesses a DNA binding constant slightly greater than that of luzopeptin A (K = 3.4 × 107 M–1vs. 1.38 × 107 M–1), with the largest share of the affinity attributed to the cyclic decadepsipeptide alone (ΔG° = –6.0 kcal mol–1) while the sequential addition of chromophores increases this value by 3.2 kcal mol–1 (for one chromophore ) and 1 kcal mol–1 (for two), respectively.12,102 Studies have shown that sandramycin unwinds DNA at a concentration similar to the highly potent luzopeptin C.12 While DNase I footprinting studies revealed that sandramycin and luzopeptin A bind preferentially to the regions containing 5′-AT residues, sandramycin has overall a higher selectivity than luzopeptin A (saturated binding at a 1 : 6.7 vs. 1 : 4.5 agent/base-pair ratio) and is also able to bisintercalate at lower concentrations. It has also been suggested that sandramycin could demonstrate two modes of binding, namely high affinity bisintercalation and lower affinity monointercalation.12
5.2 The luzopeptins
In 1980, Ohkuma and co-workers reported the laboratory isolation of a complex of six anti-tumour antibiotics, 928 A–F, from the culture broth of the aerobic strain of Actinomadura luzonensis nov. sp. (actinomycetes no. G455-101).107,108 Preparative counter-current distribution purification of the crude extract resulted in the separation of luzopeptin C8 from the rest of the mixture. Further purification by column chromatography on silica gel (ethyl acetate then 3% methanol in ethyl acetate) and chloroform–methanol crystallisation performed on both BBM 928 lots provided luzopeptin (BBM 928) A (6, 988 mg), B (7, 420 mg), C (8, 848 mg), D (130 mg), E (119 mg), and F (114 mg) as colourless crystals readily soluble in chloroform and dichloromethane.
The chromophore -bearing decadepsipetide nature of luzopeptins A, B and C was initially established using UV, IR and NMR experiments, derivatisation studies and microanalysis.107,108 The molecular formulae of luzopeptins A, B and C were established as C64H78N14O24, C62H76N14O23 and C60H74N14O22 respectively, by microanalysis and osmometric molecular weight determination.109,110 The symmetrical nature of luzopeptins A and C was deduced from NMR spectral data showing half the number of proton and carbon atoms expected from the molecular formula, a feature not found for luzopeptin B.109 Analysis of the fragments obtained by hydrolysis studies of luzopeptin C revealed the presence of D-Ser, Sar and Gly along with the unusual trans-(3S,4S)-4-hydroxy-2,3,4,5-tetrahydropyridazine-3-carboxylic acid and L-β-hydroxy-N-methylvaline, as well as their sequence in the natural product. The nature of the 3-hydroxy-6-methoxyquinaldic acid chromophore was also established by acid hydrolysis.109 Analysis of the NMR spectra of luzopeptins A, B and C and their corresponding acetylated derivatives indicated that B and A were mono- and diacetyl analogues of C, respectively, with the acetylation taking place on the hydroxyl group of the tetrahydropyridazine moiety.107,110
X-Ray crystallography and NMR studies demonstrated that the luzopeptins have a right-handed twisted rectangular β-sheet conformation stabilised by transannular hydrogen bonds between opposing Gly NH and carbonyl groups, thus generating a cis-configuration between the two chromophores with respect to the peptide ring.111,112
The luzopeptins and quinoxopeptins, which contain an identical cyclic core, differ only in their chromophores and the nature of the unusual (3S,4S)-4-hydroxy-2,3,4,5-tetrahydropyridazine-3-carboxylic acid moiety (L-Htp) acyl susbstituents. Consequently, a flexible approach allowing access to both structures has been a common aim of all synthetic routes. Such a strategy involves introducing the chromophores and functionalising the L-Htp residue late in the synthesis. As both antibiotics have a C2 symmetry axis, assembly of the central core can be readily achieved by lactamisation at the secondary amide site of two pentadepsipetide subunits.
The assembly of the D-Ser-L-Htp moiety is a key step for the synthesis. All routes disclosed to date have relied on an amino hydrazine–aldehyde intramolecular cyclisation. Stereochemical control of the corresponding α-hydrazo-β-hydroxyester precursor relied on different approaches. Quantitative asymmetric reduction of a β-ketoester 39 (Scheme 2) to the corresponding β-hydroxyester 40 proceeded with >99% ee using 0.3% Ru-(S)-Biphemp-Br2 as catalyst in the presence of H2 (6 bar, 80 °C). This was followed by a highly diastereoselective electrophilic amination in the α-position using LDA and DBAD in the presence of methyl zinc bromide (53%, >98% de), which provided, after silylation of the alcohol 41, the required hydrazino precursor.113 Further homologation of the functionalities, including generation of the aldehyde by ozonolysis, deprotection of the amino moieties and subsequent cyclisation, provided the required L-Htp moiety 42 in 50% yield over two steps. An alternative strategy includes the installation of the appropriate stereochemistry at the α- and β-positions by Sharpless asymmetric epoxidation (81%, >97% ee) of an allylic alcohol 43, homologation of the alcohol functionality of 44 to the corresponding potassium carboxylate salt and subsequent regioselective ring opening of the epoxide with hydrazine.114 Exposure of the resulting intermediate 45 to moderately acidic conditions provided the expected cyclic product 46 in a 65% yield over three steps.
 | ||
| Scheme 2 Top: Genêt’s approach to the luzopeptins.113 Bottom: Clardy’s approach to the luzopeptins.114Reagents and conditions: (a) CH3OH, H2, 0.3% Ru-(S)-Biphemp-Br2, 6 bar, 80 °C, 30 min, quantitative, 99% ee; (b) MeZnBr, LDA, DBAD, 53%, 98% de; (c) TBDMSOTf, 2,6-lutidine, 85%; (d) O3, Me2S; (e) TFA, CH2Cl2 then H2O, CH3OH, 50% over two steps; (f) tBuOOH, Ti(OiPr)4, L-(+)-diethyl tartrate, 81%; (g) RuO4; (h) CH2N2, 70% over two steps, 97% ee; (i) K2CO3, CH3OH, H2O; (j) NH2NH2, H2O; (k) TFA, H2O, 65% over three steps. | ||
Ciufolini and co-workers assembled the α-hydrazo-β-hydroxyester intermediate 49via Bakers' yeast reduction of a 5-benzyloxy-β-ketoester derived from 47 to the corresponding β-hydroxyester (48, 89% ee) (Scheme 3). Subsequent oxidation of the primary alcohol to the aldehyde and introduction of the hydrazine moiety using Gennari–Evans–Vederas conditions (61%, 95% de) gave the target 49.115,116 With the total synthesis in mind, the D-Ser moiety was installed in a protected form prior to pyridazine formation. Cyclisation was performed in 9 : 1 TFA–H2O to give 50 in a 97% yield.
 | ||
Scheme 3 Top: Cuifolini’s approach to the luzopeptins.115,116 Bottom: Boger’s approach to the luzopeptins.117Reagents and conditions: (a) NaH, BuLi, BnOCH2Cl; (b) yeast, sucrose, 89% ee; (c) TBSCl, ImH, DMF, rt, 16 h, 90%; (d) H2, 10% Pd/C, cyclohexane, rt, 95%; (e) DMSO, (COCl)2, –78 °C, Et3N, CH2Cl2, 92%; (f) ethylene glycol, cat. PPTS, benzene, reflux, 3 h, 88%; (g) TBAF, THF, rt, 4 h, 85%; (h) Cbz–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–Cbz, LDA, THF, –78 °C, 61%; (i) Ac2O, pyridine, rt, 16 h, 95%; (j) Boc2O, H2, 10% Pd/C, rt, 8 h, 97%; (k) (R)-3-acetyl-2-oxo-oxazolidine-4-carbonyl chloride, sym-collidine, CH2Cl2, 0 °C, 0.5 h, 60%; (l) 9 : 1 TFA–H2O, 0.5 h, 97%; (m) NH2NH2, CH3CN, rt, 80%; (n) NaOH, THF–H2O, rt, 90%; (o) BuNH2, HBTU, DMF, rt, 70%; (p) Ac2O, pyridine, rt, 3 h, 95%; (q) Boc2O, cat. DMAP, Et3N, CH2Cl2, 0 °C, 15 min, 100%; (r) Cs2CO3, CH3OH, rt, 15 min, 90%; (s) AD-mix-α, 80%, 99% ee; (t) NosCl, 68%; (u) NaN3, 87%; (v) PPh3, 93%; (w) TBDMSOTf, 98%; (x) 3-(4-cyanophenyl)oxaziridine-2-carboxylic acid tert-butyl ester, DMF, 72%; (y) (R)-2-oxo-3-(2-trimethylsilanylethanesulfonyl)oxazolidine-4-carbonyl chloride, 85%; (z) Cs2CO3, CH3OH, 64%; (aa) 10% H2O–TFA, 86%. N–Cbz, LDA, THF, –78 °C, 61%; (i) Ac2O, pyridine, rt, 16 h, 95%; (j) Boc2O, H2, 10% Pd/C, rt, 8 h, 97%; (k) (R)-3-acetyl-2-oxo-oxazolidine-4-carbonyl chloride, sym-collidine, CH2Cl2, 0 °C, 0.5 h, 60%; (l) 9 : 1 TFA–H2O, 0.5 h, 97%; (m) NH2NH2, CH3CN, rt, 80%; (n) NaOH, THF–H2O, rt, 90%; (o) BuNH2, HBTU, DMF, rt, 70%; (p) Ac2O, pyridine, rt, 3 h, 95%; (q) Boc2O, cat. DMAP, Et3N, CH2Cl2, 0 °C, 15 min, 100%; (r) Cs2CO3, CH3OH, rt, 15 min, 90%; (s) AD-mix-α, 80%, 99% ee; (t) NosCl, 68%; (u) NaN3, 87%; (v) PPh3, 93%; (w) TBDMSOTf, 98%; (x) 3-(4-cyanophenyl)oxaziridine-2-carboxylic acid tert-butyl ester, DMF, 72%; (y) (R)-2-oxo-3-(2-trimethylsilanylethanesulfonyl)oxazolidine-4-carbonyl chloride, 85%; (z) Cs2CO3, CH3OH, 64%; (aa) 10% H2O–TFA, 86%. | ||
Boger et al. generated the asymmetric centres at positions 2 and 3 of an α,β-dehydro ester 51 by Sharpless dihydroxylation (80%, >99% ee).117 Selective activation of the α-hydroxyl followed by azide displacement gave 52. Staudinger reduction, reaction with an oxaziridine and protection resulted in the formation of the required α-hydrazo-β-hydroxyester. Coupling with a protected version of the D-Ser moiety to give 53 was performed prior to cyclisation (Scheme 3).
Boger’s total synthesis of the luzopeptins continued with assembly of the unusual L-β-hydroxy-N-methylvaline amino acid. NaH-promoted intramolecular ring opening of 55 provided 56 after rearrangement (Scheme 4).118 Further oxazolidinone ring opening and subsequent EDC/HOAt-promoted Boc-Gly-Sar-OH coupling afforded 57. The key pentadepsipeptide 59 was synthesised without racemisation by condensation of 57 with D-Ser-L-Htp dipeptide 58. Selective deprotection of the terminal amino and carboxylic functionalities provided respectively 60 and 61. Coupling of these subunits using EDC/HOAt led to linear peptide 62. Base-free macrocyclisation of 62, followed by in situtetrahydropyridazine formation and chromophore coupling, provided luzopeptin C (80% yield). Derivatisation of luzopeptin C (Ac2O–Py, Na2CO3) provided luzopeptin A (50% yield) and luzopeptin B (20% yield).
 | ||
| Scheme 4 Top: Boger’s total synthesis of the luzopeptins. Bottom: Ciofulini’s synthesis of the luzopeptins. Reagents and conditions: (a) Sharpless epoxidation; (b) CH3CNO, 94%; (c) NaH, 66–85%; (d) PPTs, DHP, 99%; (e) KOH, 94%; (f) Boc-Gly-Sar-OH, EDC, HOAt, 83%; (g) TsOH, 84%; (h) RuO2, NaIO4, 87%; (i) HCl, FmocCl, 79%; (j) DCC, DMAP, –20 °C to 0 °C, 73%, 56; (k) Et2NH, 100%; (l) H2, Pd/C, 78%; (m) EDC, HOAt, 64%; (n) HCO2NH4, Pd/C, 98%; (o) EDC, HOAt, 0 °C, 66%; (p) TFA, CH2Cl2, anisole, 68%; (q) i) HF, ii) 3-hydroxy-6-methoxyquinoline-2-carboxylic acid, EDC, HOBt, NaHCO3, 80%; (r) Ac2O–pyridine, Na2CO3; (s) Boc2O, THF, NaOH (10%); (t) CH2N2; (u) DHP, PPTS, CH2Cl2, 96% over three steps; (v) CH3MgBr, ether, –20 °C; (w) NaH, THF, CH3I; (x) KOH, (CH2OH)2–H2O; (y) Boc2O, CH2Cl2, rt, 99%; (z) TsOH, CH3OH, 95%; (aa) Swern oxidation; (bb) NaClO2, tBuOH, 2-methyl-2-butene, NaHPO4; (cc) allyl bromide, Et3N, 78% over three steps; (dd) TFA, CH2Cl2, 96%; (ee) BOP-Cl, CH2Cl2, 64, 63%; (ff) Pd(PPh3)4, dimedone, 80–90%; (gg) DCC, DMAP, 67, 60%; (hh) Pd(PPh3)4, dimedone; (ii) PPh3, H2O; (jj) EDC, HOAt, CH2Cl2, 26% over three steps; (kk) TFA, CH2Cl2; (ll) 3-hydroxy-6-methoxyquinaldic acid, EDC, HOBt, 55% over two steps; (mm) Py·HF, CH2Cl2, 12%. | ||
Ciufolini and colleagues also tackled the total synthesis of the luzopeptins.119 Assembly of the L-β-hydroxy-N-methylvaline amino acid was achieved using D-Ser as a template by conversion of the carboxylic acid functionality to the corresponding dimethyl alcohol, alkylation of the amino group and oxidation of the primary alcohol to the carboxylic acid.120 Allyl protection of the carboxylic acid and coupling of the resulting product 63 with N3-Gly-Sar-OH 64 using BOP-Cl yielded the key tripeptide 65 ready for later assembly of the pentadepsipeptide.121 Coupling with the readily cyclised D-Ser-L-Htp subunit 67 was achieved in a 60% yield using DCC/DMAP conditions. Simultaneous deprotection of both ends of the resulting pentadepsipeptide and treatment with EDC/HOAt provided the cyclic core of luzopeptins 68 in one pot (26% yield). Deprotection of the D-Ser amino groups and subsequent linkage of the chromophores provided luzopeptin C, after deprotection of the hydroxy functionalities of the tetrahydropyridazine residue.
In a similar fashion, Ciufolini also reported the first total synthesis of luzopeptin E2.122
Even though they are 16- to 24-fold less active than echinomycin, luzopeptins A, B, C and D all exert relatively strong in vitro anti-microbial activities against Gram-positive Bacillus subtilis (MIC 0.4 to 6.3 µg mL–1), Staphylococcus aureus (MIC 0.2 to 6.3 µg mL–1) and Streptococcus pyogenes (MIC 0.1 to 0.8 µg mL–1) with activities decreasing in the order A, D, B and C. Luzopeptins A, B and C failed to demonstrate any ability to induce prophage in lysogenic bacterium (ILB) below 100 µg mL–1.107 When tested in vitro against Plasmodium falciparum, Luzopeptin A demonstrated an LD50 against the parasite at a concentration of 9 × 10–11 M.123
As an anti-tumour agent against P388 leukaemia, luzopeptin A proved to be three times more potent than echinomycin and luzopeptin B, while luzopeptin C did not show any activity.107 When tested against a number of tumour systems, including melanoma B16 and sarcoma 180, luzopeptin A was more active than mitomycin C. Both anti-bacterial and anti-tumour activities of the BBM 928 components mirror the degree of acetylation of the cyclic peptide.107 Toxicity studies showed that luzopeptin A was slightly less toxic than echinomycin (LD50 0.13 vs. 0.11 mg kg–1 day–1) but slightly more toxic than luzopeptin B (LD50 0.18 mg kg–1 day–1) and 55 times more than mitomycin C (LD50 7.1 mg kg–1 day–1).107 However, luzopeptin A was devoid of myelotoxicity, nephrotoxicity and hepatotoxicity.124
All three luzopeptins A, B and C are excellent reverse transcriptase inhibitors at concentrations of 10 or 40 µg mL–1 (89–100% inhibition).125 When tested against HIVHTLV-III in MT-4 cells, luzopeptin C suppressed the replication of HIV (2.5–5.0 µg mL–1) without significantly affecting the viability of the host cells. When tested against HIV-1 and HIV-2 RT, luzopeptin A was a potent inhibitor, with IC50 values of 7 and 68 nM respectively.126,127 In contrast, triostin A and echinomycin were inactive against reverse transcriptase.
Luzopeptin A interacts with DNA with high affinity, as shown by quenching studies with circular superhelical PM2 DNA, giving an apparent association constant (K) of 1.93 × 107 M–1 with one molecule of bound drug for every 5–6 base-pairs at saturation.128 Interestingly, a comparative study at a low drug/DNA ratio (<0.12) showed luzopeptin C to induce stronger DNA binding than luzopeptin B (K = 9.85 × 107 M–1vs. 7.73 × 107 M–1) and A (5.49 × 107 M–1).129 No single- or double-strand DNA breaks have been observed in vitro with any of the luzopeptins.130
Bisintercalation of the chromophores was suggested by viscometric studies of the luzopeptin A–DNA complex, with DNA unwinding occurring at drug concentrations lower than that with ethidium bromide (rc = 0.054 vs. 0.091) and similar to that observed for echinomycin (rc = 0.044) with an unwinding angle θ = 43° (θ = 26° with ethidium bromide).128 Similarly, paralleling the properties of echinomycin, luzopeptin A treatment of linear rodlike calf thymus DNA resulted in a 1.5-fold increase of DNA helix length compared to ethidium bromide treatment (1.8-fold increase with echinomycin).127 When compared against each other, luzopeptin C completely relaxed DNA at a lower concentration than luzopeptin B (rc = 0.046 vs. 0.048) and luzopeptin A (rc = 0.049). As a result of this study, the unwinding angles of luzopeptins C and B were established as θ = 45.7° and 43.9° respectively.129
Although the mode of intercalation of luzopeptin A is mainly based on an intramolecular DNA binding of the two quinoline chromophores, at lower concentration (luzopeptin A/DNA < 0.18) the formation of less stable intermolecular cross-links has also been suggested using agarose gel electrophoresis techniques.131 Luzopeptins B and C also had similar interactions with DNA, and at 5- to 10-fold lower concentrations in the case of luzopeptin C. The possibility of such binding has been questioned but the use of atomic force microscopy (AFM) tends to support its existence.132,133
DNase I footprinting studies revealed that the luzopeptins do not have a rigid sequence selectivity but bind best to regions rich in 5′-AT residues.134 NMR data for the luzopeptin A–d(GCATGC)2 complex showed the drug intercalating in the DNA helix minor groove by insertion of its quinoline chromophores on each side of the central AT base-pairs (1–1.2 ppm upfield shift of 5-G and 4-T imino protons after intercalation).135 NOESY studies also suggested that the position of the drug chromophores in the groove was optimal to create stacking interactions with the purine bases on both strands in positions A2 and G4.132,136 Such selectivity and intercalation of the drug seems to arise from conformational modifications of luzopeptin going from the crystalline state to association with DNA. A break in the transannular Gly–Gly interaction results in a cis conformation of Pyr–Gly and Gly–Sar peptide bonds. This new conformation allows recognition of the 5′-AT residues by creation of a stabilising hydrogen bond between the peptide-NH Gly residue and the DNA thymine 2-carbonyl functionality. A second hydrogen bond between the peptide L-β-hydroxy-N-methylvaline residue and the DNA guanine 2-amino group also benefits from this conformational change. Despite playing a key role in the sequence recognition of echinomycin, this latter interaction only contributes to a minor extent to the formation of a stable luzopeptin–DNA complex. This was determined by binding studies of luzopeptins A, B and C to DNA sequences differing by the presence or absence of a 2-amino group on guanine and adenine residues.137 Van der Waals interactions between the methyl groups of luzopeptin and the minor groove surface have also been identified.133 All the base-pairs retain a Watson–Crick alignment, with no evidence of Hoogsteen base-pairing.
Luzopeptins A, B and C did not show any inhibition of protein synthesis. The inhibitory effects of the luzopeptins on DNA synthesis depend on the complexity of the biological entity.138 Luzopeptins A–C displayed similar strong inhibitory effects on Micrococcus lysodeikticus DNA polymerase activity in vitro. However, luzopeptin C proved to be two times less effective than luzopeptins A and B when tested against [3H]thymidine incorporation in isolated nuclei of Novikoff hepatoma cells, and was totally ineffective in the parent hepatoma cell line (IC50 150 nM for luzopeptin A, and 4–6 µM for luzopeptin B).138
Luzopeptin C is an extremely potent inhibitor of isolated E. coli RNA polymerase activity in vitro, is a better inhibitor than luzopeptin B in isolated nuclei (IC50 0.08 µg per 106 nuclei vs. 0.33 µg per 106 nuclei) and is 20 times more effective than luzopeptin A (IC50 0.21 µg per 106 nuclei). However, luzopeptin C is ineffective in intact cells while A and B have IC50 values of 30 nM and 3.7 µM respectively. Based on these results, it has been suggested that the cytotoxicity and anti-tumour activities of the luzopeptins are dependent on the ability of these drugs to traverse the cell membrane, and that the level of acetylation of the tetrahydropyridazine is a key structural feature.138
5.3 The quinoxapeptins
Quinoxapeptins A 9 and quinoxapeptin B10 were isolated from a Betula papyrifera bark disk nocardioform actinomycete.126 Separation of the two quinoxapeptins was achieved by preparative TLC to provide the two homologues A and B as white solids. They are cyclic chromodecadepsipeptides that share a similar peptide backbone with the luzopeptins but differ in the nature of their chromophores and acyl substituents attached to the tetrahydropyridazine-3-carboxylic acid residue.12 The quinoxapeptin chromophore is based upon 6-methoxyquinoxaline-2-carboxylic acid, while the acetyl group of the luzopeptins is replaced by 2-methylcyclopropane carboxylic acid. Interestingly, while quinoxapeptin A exhibits a C2 symmetry axis, the quinoxapeptin Btetrahydropyridazine residues are non-equivalent, as one bears an acetyl functionality and the other a methylcyclopropane. The total synthesis of the quinoxapeptins by Boger and co-workers resulted in elucidation of the 1S,2S absolute stereochemistry of the cyclopropane moiety and confirmation of the relative and absolute configuration of the cyclic decadepsipeptide.139 Full characterisation of both quinoxapeptins A and B was reported in this work.
To date only one total synthesis of the quinoxapeptins has been reported.139 Boger and co-workers designed a synthetic pathway that would give access to both luzopeptins and quinoxapeptins from a common intermediate that could be functionalised in the final steps of the synthesis. Therefore, as reported in the total synthesis of luzopeptins, in situ assembly of the tetrahydropyridazine residues under acidic conditions (68% yield), followed by coupling of 6-methoxyquinoxaline-2-carboxylic acid chromophores with the deprotected D-Ser amino groups, was performed in a 65% yield over two steps to provide the unnatural quinoxapeptin C11. With this synthetic intermediate in hand, acetylation using (S,S)-2-methylcyclopropanecarboxylic acid gave quinoxapeptin A in 80% yield along with 18% of monoacetylated product, which was treated with Ac2O to provide quinoxapeptin B in 62% yield.139
Both quinoxapeptins A and B are potent inhibitors of HIV reverse transcriptase (RT). Quinoxapeptin A showed the most potency, with IC50 values of 4 and 40 nM against HIV-1 and HIV-2 enzymes respectively (quinoxapeptin B: IC50 10 nM against HIV-1 and 100 nM against HIV-2).126Quinoxapeptin A was identified as a non-competitive primer-template substrate that inhibits either single or double mutant RT with the same ability (IC50 8–12 and 6 nM, respectively). It has also been shown to bind reversibly to the enzyme. Its weaker mammalian DNA polymerase inhibition (IC50 2563–494 nM) enhanced the remarkable specificity of quinoxapeptin A towards HIV RT. In the total synthesis of the quinoxapeptins, Boger et al. made a non-natural analogue 11, devoid of acyl substituents on the tetrahydropyridazine residue, named quinoxapeptin C by analogy with the luzopeptins. Studies on this derivative established that it was more effective against HIV-1 RT (0.3 µM) than homologues A and B (0.6 vs. 0.9 µM), but was also less cytotoxic against L1210 (>100 nM) than A and B (0.3 and 2 nM).12
Quinoxapeptin A binds with a higher affinity for duplex DNA than B (K = 1.51 × 107 M–1vs. 1.04 × 107 M–1) and non-natural quinoxapeptin C (0.43 × 107 M–1) Both quinoxapeptins A and C had similar minor increased affinities for 5′-TA intercalation sites.12
5.4 Quinaldopeptin
Quinaldopeptin 12 was first isolated from the culture broth of Streptoverticillium album (actinomycetes strain Q132-6) along with aureothricin and a methylpentene.140 It has also been isolated as a co-metabolite from a culture broth of actinomyces strain A499.141 The structure of quinaldopeptin was established using UV, IR and NMR experiments, derivatisation studies and microanalysis. The molecular formula was established as C62H78N14O14 by microanalysis.140 NMR experiments were performed on a more soluble diacetyl derivative. The symmetrical nature of the natural product was deduced by the NMR spectra showing half the number of proton and carbon atoms expected from the molecular formula. The IR spectrum showed the presence of amide bonds (1690–1530 cm–1) and a lack of ester linkages. UV spectra similar to that of sandramycin indicated identical chromophores, i.e. 3-hydroxyquinaldic acids.140 Fragment analysis from acidic hydrolysis (6 M HCl) revealed the presence of 3-hydroxyquinaldic acid, Gly, Sar, L-pipecolic acid and D-erythro-α,β-diaminobutyric acid as constituents of the natural product. Partial degradation studies (3 M HCl) established the natural product full amino acid sequence and showed that four L-pipecolic acid residues were present in the central core.140Quinaldopeptin has a strong activity against aerobic Gram-positive bacteria [S. aureus (MIC 0.4 µg mL–1), S. faecalis (MIC 1.6 µg mL–1), S. pyogenes (MIC 0.2 µg mL–1), B. subtilis (MIC 0.4 µg mL–1), P. subtilis (MIC 100 µg mL–1)] and anaerobic Gram-positive bacteria [C. difficile (MIC 0.4 µg mL–1)], but was less active against Gram-negative bacteria. It also showed a positive response to the fungus Cryptococcus neoformans (MIC 3.1 µg mL–1). Parallel testing of the diacetyl derivative provided similar data.140
When tested in vitro against melanoma B16 and Moser cells, quinaldopeptin had good activity (IC50 0.0007 and 0.04 µg mL–1 respectively) and was more potent than chromomycin A3 against B16 melanoma (IC50 0.002 µg mL–1). In vivo anti-tumour activity tested against lymphocytic leukaemia P388 in mice indicated greater potency than mitomycin C (MED 0.3 mg kg–1 day–1).140
6 Conclusions
The bisintercalator natural products are high affinity DNA binding ligands with potential as tools for chemical biology studies as well as for therapeutic use in anti-bacterial, anti-viral and anti-cancer therapies. New compounds continue to appear, with one of the newest, the marine natural product thiocoraline, under development and moving towards clinical trials. In addition, the interesting structures, variation in sequence selectivity and spectrum of activity of these compounds means that they will continue to interest synthetic and natural products chemists, as well as chemical biologists, for some time to come.7 Acknowledgements
This article is written in recognition of the retirement of Professor Michael Waring, one of the fathers of this area of research. Work in the Searcey and Malkinson groups is supported by the EPSRC, the Association for International Cancer Research, the BBSRC and the School of Pharmacy. We thank our colleagues Michael Anim and Salvatore Cipolla, who have also contributed to our research in this area, and our collaborators Keith Fox and Donald Fitzmaurice. As this is a large area, we apologise in advance to any group that feels their work was not represented.8 References
- R. Corbaz, L. Ettlinger, E. Gaumann, W. Keller-Schierlein, F. Kradolfer, L. Neipp, V. Prelog, P. Reusser and H. Zahner, Helv. Chim. Acta, 1957, 40, 199 CrossRef CAS.
- T. Yoshida, K. Katagiri and S. Yokozawa, J. Antibiot., 1961, 14(ser. A), 330 CAS.
- W. Keller-Schierlein, M. Mihailovic and V. Prelog, Helv. Chim. Acta, 1959, 42, 305 CrossRef CAS.
- A. Dell, D. H. Williams, H. R. Morris, G. A. Smith, J. Feeney and G. C. K. Roberts, J. Am. Chem. Soc., 1975, 97, 2497 CrossRef CAS.
- Y. S. Park, Y. H. Kim, S. K. Kim and S. J. Choi, Bioorg. Med. Chem. Lett., 1998, 8, 731 CrossRef CAS.
- Y. B. Kim, Y. H. Kim, J. Y. Park and S. K. Kim, Bioorg. Med. Chem. Lett., 2004, 14, 541 CrossRef CAS.
- A. B. Khan, A. P. Bhaduri, C. M. Gupta and M. M. Dhar, Indian J. Biochem., 1969, 6, 220 Search PubMed.
- J. Arif, C. Singh, A. P. Bhaduri, C. M. Gupta, A. W. Khan and M. M. Dhar, Indian J. Biochem., 1970, 7, 193 Search PubMed.
- D. Gauvreau and M. J. Waring, Can. J. Microbiol., 1984, 30, 721 CAS.
- D. Gauvreau and M. J. Waring, Can. J. Microbiol., 1984, 30, 730 CAS.
- K. R. Fox, D. Gauvreau, D. C. Goodwin and M. J. Waring, Biochem. J., 1980, 191, 729 CAS.
- D. L. Boger, M. W. Ledeboer, M. Kume, M. Searcey and Q. Jin, J. Am. Chem. Soc., 1999, 121, 11375 CrossRef CAS.
- A. Cornish, M. J. Waring and R. D. Nolan, J. Antibiot., 1983, 36, 1664 CAS.
- D. G. Reid, D. M. Doddrell, D. H. Williams and K. R. Fox, Biochim. Biophys. Acta, 1984, 798, 111 CrossRef CAS.
- G. Schmoock, F. Pfennig, J. Jewiarz, W. Schlumbohm, W. Laubinger, F. Schauwecker and U. Keller, J. Biol. Chem., 2005, 280, 4339 CAS.
- M. J. Waring and L. P. G. Wakelin, Nature, 1974, 252, 653 CAS.
- G. Ughetto, A. H. J. Wang, G. J. Quigley, G. A. Vandermarel, J. H. Vanboom and A. Rich, Nucleic Acids Res., 1985, 13, 2305.
- D. E. Gilbert and J. Feigon, Biochemistry, 1991, 30, 2483 CrossRef CAS.
- L. P. G. Wakelin, Med. Res. Rev., 1986, 6, 275 CAS.
- C. Bailly and M. J. Waring, J. Biomol. Struct. Dyn., 1995, 12, 869 Search PubMed.
- G. G. Gause, N. P. Loshkareva and I. B. Zbarsky, Biochim. Biophys. Acta, 1968, 166, 752.
- L. P. G. Wakelin and M. J. Waring, Biochem. J., 1976, 157, 721 CAS.
- C. M. L. Low, H. R. Drew and M. J. Waring, Nucleic Acids Res., 1984, 12, 4865 CAS.
- C. Jeppesen and P. E. Nielsen, Eur. J. Biochem., 1989, 182, 437 CrossRef CAS.
- M. M. Vandyke and P. B. Dervan, Science, 1984, 225, 1122 CrossRef CAS.
- M. Searcey, S. McClean, B. Madden and L. P. G. Wakelin, J. Chem. Soc., Perkin Trans. 2, 1997, 523–532 RSC.
- K. D. Leslie and K. R. Fox, Biochemistry, 2002, 41, 3484 CrossRef CAS.
- M. C. Fletcher and K. R. Fox, Biochemistry, 1996, 35, 1064 CrossRef CAS.
- K. R. Fox and M. J. Waring, Biochemistry, 1984, 23, 2627 CrossRef CAS.
- D. Mendel and P. B. Dervan, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 910 CrossRef CAS.
- C. Jeppesen and P. E. Nielsen, FEBS Lett., 1988, 231, 172 CrossRef CAS.
- M. J. McClean, F. J. Seela and M. J. Waring, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9687 CrossRef.
- E. Gilbert, G. A. van der Marel, J. H. van Boom and J. Feigon, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 3006 CrossRef.
- J. A. Cuesta-Seijo and G. M. Sheldrick, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 442 CrossRef.
- D. E. Gilbert and J. Feigon, Biochemistry, 1991, 30, 2483 CrossRef CAS.
- X. L. Gao and D. J. Patel, Biochemistry, 1988, 27, 1744 CrossRef CAS.
- F. Leng, J. B. Chaires and M. J. Waring, Nucleic Acids Res., 2003, 31, 6191 CrossRef CAS.
- J. H. Harvey, M. Mcfadden, W. G. Andrews, P. J. Byrne, J. D. Ahlgren and P. V. Woolley, Cancer Treat. Rep., 1985, 69, 1365 Search PubMed.
- B. Muss, J. A. Blessing and J. Malfetano, Am. J. Clin. Oncol., 1990, 13, 191.
- B. Muss, J. A. Blessing, V. V. Baker, D. R. Barnhill and M. D. Adelson, Am. J. Clin. Oncol., 1990, 13, 299.
- S. A. Taylor, B. Metch, S. P. Balcerzak and K. H. Hanson, Invest. New Drugs, 1990, 8, 381 CAS.
- R. L. Schilsky, D. Faraggi, A. Korzun, N. Vogelzang, J. Ellerton, W. Wood and I. C. Henderson, Invest. New Drugs, 1991, 9, 269 CAS.
- S. A. Taylor, J. Crowley, J. Townsend, F. S. Vogel, H. Eyre, J. J. Braun and J. W. Goodwin, J. Neuro-Oncol., 1993, 15, 181 CrossRef CAS.
- M. Kuroya, N. Ishida, K. Katagiri, T. Shoji, T. Yoshida, M. Mayama, K. Sato, S. Matsuura, Y. Niinome and O. Shiratori, J. Antibiot., 1961, 14, 325.
- J. Shoji and K. Katagiri, J. Antibiot., 1961, A14, 335.
- H. Otsuka and J. Shoji, J. Antibiot., 1963, A16, 52.
- H. Otsuka and J. Shoji, Tetrahedron, 1965, 21, 2931 CrossRef CAS.
- J. Shoji, K. Toria and H. Otsuka, J. Org. Chem., 1965, 30, 2772 CrossRef CAS.
- H. Otsuka and J. Shoji, J. Antibiot., 1976, 29, 107 CAS.
- H. Otsuka and J. Shojii, J. Antibiot., 1965, A18, 134.
- H. C. Koppel, I. L. Honigberg, R. H. Springer and C. C. Cheng, J. Org. Chem., 1963, 28, 1119 CrossRef CAS.
- S. Gerchakov, P. J. Whitman and H. P. Schultz, J. Med. Chem., 1966, 9, 266 CrossRef CAS.
- S. Gerchakov and H. P. Schultz, J. Med. Chem., 1969, 12, 141 CrossRef CAS.
- T. L. Ciardelli and R. K. Olsen, J. Am. Chem. Soc., 1977, 99, 2807 CrossRef.
- M. A. Viswamitra, O. Kennard, W. Cruse, E. Egert, G. Sheldrick, P. Jones, M. J. Waring, L. P. G. Wakelin and R. K. Olsen, Nature, 1981, 289, 817 CrossRef CAS.
- T. L. Ciardelli, P. K. Chakravarty and R. K. Olsen, J. Am. Chem. Soc., 1978, 100, 7684 CrossRef CAS.
- M. K. Dhaon, J. H. Gardner and R. K. Olsen, Tetrahedron, 1982, 38, 57 CrossRef CAS.
- P. K. Chakravarty and R. K. Olsen, Tetrahedron Lett., 1978, 19, 1613 CrossRef.
- M. Shin, H. Otsuka and K. Inouye, Bull. Chem. Soc. Jpn., 1984, 57, 2203 CrossRef CAS.
- N. Higuchi, Y. Kyogoku, M. Shin and K. Inouye, Int. J. Pept. Protein Res., 1983, 21, 541 CAS.
- M. Shin, K. Inouye and N. Higuchi, Bull. Chem. Soc. Jpn., 1984, 57, 2211 CrossRef CAS.
- R. K. Olsen, K. Ramasamy, K. L. Bhat, C. M. L. Low and M. J. Waring, J. Am. Chem. Soc., 1986, 108, 6032 CrossRef CAS.
- A. Hassner and V. Alexanian, Tetrahedron Lett., 1978, 19, 4475 CrossRef.
- D. L. Boger and J. K. Lee, J. Org. Chem., 2000, 65, 5996 CrossRef CAS.
- J. P. Malkinson, M. K. Anim, M. Zloh, M. Searcey, A. J. Hampshire and K. R. Fox, J. Org. Chem., 2005, 70, 7654 CrossRef CAS.
- B. Dietrich and U. Diederichsen, Eur. J. Org. Chem., 2005, 147 CrossRef CAS.
- T. Yoshida and K. Katagiri, Biochemistry, 1969, 6, 2645 CrossRef.
- A. Cornish, K. R. Fox and M. J. Waring, Antimicrob. Agents Chemother., 1983, 23, 221 CAS.
- A. Cornish, K. R. Fox, S. Santikarn, M. J. Waring and D. H. Williams, J. Gen. Microbiol., 1985, 131, 561 CAS.
- K. Glund, W. Schlumbohm, M. Bapat and U. Keller, Biochemistry, 1990, 29, 3522 CrossRef CAS.
- S. Matsuura, J. Antibiot., 1965, A18, 43.
- M. J. Waring, L. P. G. Wakelin and J. Lee, Biochim. Biophys. Acta, 1975, 407, 200 CAS.
- J. S. Lee and M. J. Waring, Biochem. J., 1978, 173, 115 CAS.
- C. M. L. Low, R. K. Olsen and M. J. Waring, FEBS Lett., 1984, 176, 414 CrossRef CAS.
- A. H.-J. Wang, G. Ughetto, G. J. Quigley, T. Hakoshima, G. A. van der Marel, J. H. van Boom and A. Rich, Science, 1984, 225, 1115 CrossRef CAS.
- J. S. Lee and M. J. Waring, Biochem. J., 1978, 173, 129 CAS.
- F. Takusagawa, J. Antibiot., 1985, 38, 1596 CAS.
- K. J. Addess and J. Feigon, Biochemistry, 1994, 33, 12386 CrossRef CAS.
- K. Waterloh, R. K. Olsen and K. R. Fox, Biochemistry, 1992, 31, 6246 CrossRef CAS.
- C. M. Low, K. R. Fox, R. K. Olsen and M. J. Waring, Nucleic Acids Res., 1986, 14, 2015 CAS.
- K. J. Addess, D. E. Gilbert, R. K. Olsen and J. Feigon, Biochemistry, 1992, 31, 339 CrossRef CAS.
- K. J. Addess, J. S. Sinsheimer and J. Feigon, Biochemistry, 1993, 32, 2498 CrossRef CAS.
- K. J. Addess and J. Feigon, Biochemistry, 1994, 33, 12397 CrossRef CAS.
- C. Bailly and M. J. Waring, Biochem. J., 1998, 330, 81 CAS.
- M. Lavesa and K. R. Fox, Anal. Biochem., 2001, 293, 246 CrossRef CAS.
- E. Marco, A. Negri, F. J. Luque and F. Gago, Nucleic Acids Res., 2005, 33, 6214 CrossRef CAS.
- A. Malcolm, J. R. Moffatt, K. R. Fox and M. J. Waring, Biochim. Biophys. Acta, 1982, 699, 211 CrossRef CAS.
- F. Romero, F. Espliego, J. P. Baz, T. G. de Quesada, D. Gravalos, F. de la Calle and J. L. Fernandez-Puentes, J. Antibiot., 1997, 50, 734 CAS.
- J. P. Baz, L. M. Canedo, J. L. Fernandez-Puentes and M. V. S. Elipe, J. Antibiot., 1997, 50, 738.
- D. L. Boger and S. Ichikawa, S., J. Am. Chem. Soc., 2000, 122, 2956 CrossRef CAS.
- D. L. Boger, S. Ichikawa, W. C. Tse, M. P. Hedrick and Q. Jin, J. Am. Chem. Soc., 2001, 123, 561 CrossRef CAS.
- E. Erba, D. Bergamaschi, S. Ronzoni, M. Faretta, S. Taverna, M. Bonfanti, C. V. Catapano, G. Faircloth, J. Jimeno and M. D'Incalci, Br. J. Cancer, 1999, 80, 971 CrossRef CAS.
- D. Bergamaschi, M. Faretta, S. Ronzoni, S. Taverna, P. de Feudis, M. Bonfanti, G. Guidi, G. Faircloth, J. Jimeno, M. D'Incalci and E. Erba, Eur. J. Histochem., 1997, 41(suppl. 2), 63 Search PubMed.
- (a) R. W. Sparidans, R. E. C. Henrar, J. M. Jimeno, G. Faircloth, P. Floriano and J. H. Beijnen, J. Chromatogr., B: Biomed. Sci. Appl., 1999, 726, 255 CrossRef CAS; (b) J. Yin, P. Aviles, W. Lee, C. Ly, M. J. Guillen, P. Calvo, I. Manzanares and G. Faircloth, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2003, 794, 89 CrossRef CAS.
- E. F. A. Brandon, R. W. Sparidans, I. Meijerman, I. Manzanares, J. H. Beijnen and J. H. M. Schellens, Invest. New Drugs, 2004, 22, 241 CrossRef CAS.
- H. Okada, H. Suzuki, T. Yoshinari, H. Arakawa, A. Okura, H. Suda, A. Yamada and D. Uemura, J. Antibiot., 1994, 47, 129 CAS.
- T. Yoshinari, H. Okada, A. Yamada, D. Uemura, H. Oka, H. Suda and A. Okura, Jpn. J. Cancer Res., 1994, 85, 550 CAS.
- K. Kurosawa, K. Takahashi and E. Tsuda, J. Antibiot., 2001, 54, 615 CAS.
- K. Takahashi, H. Koshino, Y. Esumi, E. Tsuda and K. Kurosawa, J. Antibiot., 2001, 54, 622 CAS.
- J. A. Matson and J. A. Bush, J. Antibiot., 1989, 42, 1763 CAS.
- J. A. Matson, K. L. Colson, G. N. Belofsky and B. B. Bleiberg, J. Antibiot., 1993, 46, 162 CAS.
- D. L. Boger, J.-H. Chen and K. W. Saionz, J. Am. Chem. Soc., 1996, 118, 1629 CrossRef CAS.
- D. L. Boger, J.-H. Chen, K. W. Saionz and Q. Jin, Bioorg. Med. Chem., 1998, 6, 85 CrossRef CAS.
- D. L. Boger and J.-H. Chen, Bioorg. Med. Chem. Lett., 1997, 7, 919 CrossRef CAS.
- D. L. Boger and K. W. Saionz, Bioorg. Med. Chem., 1999, 7, 315 CrossRef CAS.
- J. L. Leroy, X. Gao, V. Misra, M. Gueron and D. J. Patel, Biochemistry, 1992, 31, 1407 CrossRef CAS.
- H. Ohkuma, F. Sakai, Y. Nishiyama, M. Ohbayashi, H. Imanishi, M. Konishi, T. Miyaki, H. Koshiyama and H. Kawaguchi, J. Antibiot., 1980, 33, 1087 CAS.
- K. Tomita, Y. Hoshino, T. Sasahira and H. Kawaguchi, H., J. Antibiot., 1980, 33, 1098 CAS.
- M. Konishi, H. Ohkuma, F. Sakai, T. Tsuno, H. Koshiyama, T. Naito and H. Kawaguchi, J. Antibiot., 1981, 34, 148 CAS.
- M. Konishi, H. Ohkuma, F. Sakai, T. Tsuno, H. Koshiyama, T. Naito and H. Kawaguchi, J. Am. Chem. Soc., 1981, 103, 1241 CrossRef CAS.
- M. S. Searle, J. G. Hall and L. P. G. Wakelin, Biochem. J., 1988, 256, 271 CAS.
- E. Arnold and J. Clardy, J. Am. Chem. Soc., 1981, 103, 1243 CrossRef CAS.
- C. Greck, L. Bischoff and J. P. Genêt, Tetrahedron: Asymmetry, 1995, 6, 1989 CrossRef CAS.
- P. Hughes and J. Clardy, J. Org. Chem., 1989, 54, 3260 CrossRef CAS.
- M. A. Ciufolini and N. Xi, J. Org. Chem., 1997, 62, 2320 CrossRef CAS.
- M. A. Ciufolini, D. Valognes and N. Xi, J. Heterocycl. Chem., 1999, 36, 1409 CrossRef CAS.
- D. L. Boger and G. Schüle, J. Org. Chem., 1998, 63, 6421 CrossRef CAS.
- D. L. Boger, M. W. Ledeboer and M. Kume, J. Am. Chem. Soc., 1999, 121, 1098 CrossRef CAS.
- D. Valognes, P. Belmont, N. Xi and M. A. Ciufolini, Tetrahedron Lett., 2001, 42, 1907 CrossRef CAS.
- M. A. Ciufolini and S. Swaminathan, Tetrahedron Lett., 1989, 23, 3027 CrossRef.
- M. A. Ciufolini, D. Valognes and N. Xi, Tetrahedron Lett., 1999, 40, 3693 CrossRef CAS.
- M. A. Ciufolini, D. Valognes and N. Xi, Angew. Chem., Int. Ed., 2000, 39, 2493 CrossRef CAS.
- S. Lee and J. Inselburg, J. Parasitol., 1993, 79, 780 Search PubMed.
- W. C. Rose, J. E. Schurig, J. B. Huftalen and W. T. Bradner, Cancer Res., 1983, 43, 1504 CAS.
- Y. Inouye, Y. Take and S. Nakamura, J. Antibiot., 1987, 40, 100 CAS.
- R. B. Lingham, A. H. M. Hsu, J. A. O'Brien, J. M. Sigmund, M. Sanchez, M. M. Gagliardi, B. K. Heimbuch, O. Genilloud, I. Martin, M. T. Diez, C. F. Hirsch, C. D. L. Zink, J. M. Liesch, G. E. Koch, S. E. Gartner, G. M. Garrity, N. N. Tsou and G. M. Salituro, J. Antibiot., 1996, 49, 253 CAS.
- G. M. Garrity, O. Genilloud, A. H. M. Hsu, R. B. Lingham, B. Russell, I. Martin, G. M. Salituro, M. Sanchez, J. M. Sigmund and N. N. Tsou, Br. Pat. (Merck), GB-B 2294265, 1996(Chem. Abstr., 1996, 125, 112914g) Search PubMed.
- C.-H. Huang, S. Mong and S. T. Crook, Biochemistry, 1980, 19, 5537 CrossRef CAS.
- C.-H. Huang and S. T. Crooke, Cancer Res., 1985, 45, 3768 CAS.
- C.-H. Huang, A. W. Prestayko and S. T. Crooke, Biochemistry, 1982, 21, 3704 CrossRef CAS.
- C.-H. Huang, C. K. Mirabelli, S. M. Mong and S. T. Crooke, Cancer Res., 1983, 43, 2718 CAS.
- K. R. Fox and C. Wooley, Biochem. Pharmacol., 1990, 39, 941 CrossRef CAS.
- T. Berge, E. L. Haken, M. J. Waring and R. M. Henderson, J. Struct. Biol., 2003, 142, 241 CrossRef CAS.
- K. R. Fox, H. Davies, G. R. Adams, J. Portugal and M. J. Waring, Nucleic Acids Res., 1988, 16, 2489 CAS.
- M. S. Searle, J. G. Hall, W. A. Denny and L. P. G. Wakelin, Biochem. J., 1989, 259, 433 CAS.
- X. Zhang and D. J. Patel, Biochemistry, 1991, 30, 4026 CrossRef CAS.
- C. Bailly, S. Crow, A. Minnock and M. J. Waring, Nucleosides, Nucleotides Nucleic Acids, 2000, 19, 1337 CrossRef CAS.
- C.-H. Huang and S. T. Crooke, Anti-Cancer Drug Des., 1986, 1, 87 CAS.
- D. L. Boger, M. W. Ledeboer, M. Kume and Q. Jin, Angew. Chem., Int. Ed., 1999, 38, 2424 CrossRef CAS.
- S. Toda, K. Sugarawa, Y. Nishiyama, M. Ohbayashi, N. Ohkusa, H. Yamamoto, M. Konishi and T. Oki, J. Antibiot., 1990, 43, 796 CAS.
- R. W. Rickards, J. M. Rothschild and E. Lacey, J. Antibiot., 1998, 51, 1093 CAS.
Footnote |
| † Abbreviations used: Acm: acetamidomethyl; Biphemp: (6,6′-dimethylbiphenyl-2,2′-diyl)bis(diphenylphosphine); Boc: tert-butoxycarbonyl; BOP-Cl: bis(2-oxo-3-oxazolidinyl)phosphinic chloride; Cbz: benzyloxycarbonyl; CYP: cytochrome P450 ; DBAD: dibenzylazodicarboxylate; DCC: dicyclohexylcarbodiimide; DIC: diisopropylcarbodiimide; DMAP: 4-dimethylaminopyridine; Dpr: diaminopropionic acid; EDC: 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride; Fmoc: 9-fluorenylmethoxycarbonyl; HBTU: 2-(1H-benzotrizol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; HOAt: 1-hydroxy-9-azabenzotriazole; HOBt: 1-hydroxybenzotriazole; ImH: imidazole; NHS: N-hydroxysuccinimide; PPA: polyphosphoric acid; PPTS: pyridinium p-toluenesulfonate; Tce: 2,2,2-trichloroethyl. |
| This journal is © The Royal Society of Chemistry 2007 |