‘GaI’: A new reagent for chemo- and diastereoselective C–C bond forming reactions†
Shaun P.
Green
,
Cameron
Jones
*,
Andreas
Stasch
and
Richard P.
Rose
Centre for Fundamental and Applied Main Group Chemistry, School of Chemistry, Main Building, Cardiff University, Cardiff, UK CF10 3AT. E-mail: Jonesca6@cardiff.ac.uk
First published on 8th December 2006
Abstract
The potential of ‘GaI’ as a reductant in organic transformations, in particular C–C bond forming reactions involving α-functionalised ketones, has been investigated. Of most interest are the diastereoselective aldol coupling reactions of the α-alkoxy ketones, PhC(O)C(H)(OR)Ph, R = Me, Et or iPr, which give the novel γ-alkoxy, β-hydroxy ketones, PhC(O)C(H)(Ph)C(OH)(Ph)C(H)(OR)Ph, containing three contiguous stereogenic centres. The diastereostereoselectivity of these reactions was established from NMR spectroscopic and X-ray crystallographic studies of the products. When R = Me, the 2R,3S,4R/2S,3R,4S-enantiomeric pair is formed, whereas when R = Et or iPr, 2R, 3R, 4S/2S, 3S, 4R-diastereoisomers result. These differences are rationalised in terms of the likely transition states of the reactions. The same products are not formed when higher oxidation salts of gallium, or InI, are employed as the inorganic reagent. The reactivity of “GaI” towards α-halo ketones, a 1,2-diketone and α,β-unsaturated ketones has also been explored. Again, the outcomes of these reactions have been compared to those involving Ga(II) and Ga(III) reagents.
Introduction
C–C bond forming reactions are fundamental to organic synthesis and have been carried out using inorganic and organometallic reagents incorporating metals from across the periodic table. From group 13, boron(III) and aluminium(III) compounds have been amongst the widely used reagents for this purpose. More recently, indium compounds have found increasing usage in organic synthetic methodology. This can involve reagents with indium in the +3 oxidation state, but there is an escalating interest in the exploitation of indium metal and indium(I) halides as reductants in processes such as Barbier allylations, Reformatsky reactions, aldol additions and propargylation reactions.1,2 Although not as widely studied, gallium metal is beginning to be accepted as a reagent in a similar array of reactions.2,3Low oxidation state gallium halides have been almost completely neglected as reducing agents in C–C bond forming reactions. The reasons for this most likely include the fact that the former are not commercially available, and are more oxygen and moisture sensitive than their indium counterparts. Indeed, prior to our involvement in this area, it appears that only two reports4 have detailed the employment of a gallium sub-halide in organic synthesis. In these, Saigo et al. described the use of gallium(II) chloride (which actually exists as a mixed oxidation salt, GaI[GaIIICl4]) in the one-pot “reductive Friedel–Crafts” coupling reactions of carbonyl compounds or dimethylacetals with aromatics.
Our interest in low oxidation state gallium chemistry largely involves the stabilisation and coordination chemistry of gallium(I) N-heterocyclic carbene analogues.5 The most important starting material in the synthesis of such heterocycles is Green’s “GaI” which acts as a source of gallium(I) iodide.6 This insoluble green powder is straightforwardly prepared by the reaction of gallium metal with one half of an equivalent of diiodine in toluene under ultrasonic conditions (ultrasonic bath). Although it is very oxygen sensitive, it is thermally stable and can be stored indefinitely under an inert atmosphere without loss of activity. Although its formulation is not definitely known, the results of a Raman spectroscopic study suggest it predominately consists of the mixed oxidation salt, Ga2I[Ga2III6], with an average gallium oxidation state of +1.5.7
In preliminary studies, we have shown that this material is, not surprisingly, a more potent reducing reagent than InI. For example, it can facilely reductively couple diynes (to give ene-diynes)8 or bulky 2-(imino)pyridines9 that are unreactive towards the indium reagent. Given the apparent chemoselectivity of “GaI” over InI, we were keen to extend our studies into its use in C–C bond forming reactions. In this paper we detail its unusual reactivity towards α-alkoxy ketones, α-halo ketones and α-diketones, which in the case of α-alkoxy ketones, leads to the diastereoselective formation of γ-alkoxy β-hydroxy ketones with three contiguous stereogenic centres.
Results and discussion
(i) Aldol-type coupling reactions of α-alkoxy ketones
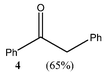
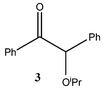
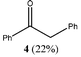
Strong reducing agents such as SmI2 have been shown to reduce α-alkoxy ketones, e.g. benzoin methyl ether, to the corresponding ketone, e.g. deoxybenzoin, under mild, neutral conditions.10 In attempts to carry out similar reductions of α-alkoxy ketones with “GaI”, deoxybenzoin, 4, was formed but unexpected C–C coupled products also resulted. The reactions of benzoin methyl ether 1, benzoin ethyl ether 2 and benzoin isopropyl ether 3, with four equivalents of “GaI” were carried out in toluene, at −78 °C, with subsequent slow warming to room temperature. These afforded the novel tri-functional aldol coupled products 1a (2R,3S,4R/2S,3R,4S-product), 2a and 3a (2R,3R,4S/2S,3S,4R-products), respectively, with complete diastereocontrol within detection limits (see Table 1).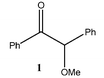






The reactions are thought to proceed via an initial reduction of the substrate to generate short-lived gallium-enolates (assumed to exist as their E-isomers), “[(RO)IGa{OC(Ph)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Ph)(H)}]”
5, which upon formation, undergo aldol-type reactions with unreacted substrate to give the observed products. NMR spectroscopic studies of the reaction mixtures prior to aqueous work-up did not show any evidence of 5 or the expected gallium alkoxide conjugate bases of 1a–3a. Instead, only 4 and the quenched products were present. This suggests that product quenching via proton abstraction from the toluene reaction solvent or another reaction component readily occurs. The increasing yield of 4 with increasing size of R could, therefore, be explained by a competition between solvent quenching of 5 (to give 4) and its nucleophilic attack on the α-alkoxy ketone substrate. The latter process would, presumably, be slowed with increasing size of the alkoxy substituent, thus favouring the formation of 4. When the reactions were repeated utilising benzene as the solvent, the resultant yields and diastereoselectivities were nearly identical to the toluene reactions. Intriguingly, when hexane was used as a solvent no reactions occurred. One explanation for this is that arene solvents are required to partly solubilise the “GaI” reagent. Indeed, it is well known that Ga(I) salts engage in π-arene interactions in solution.11
C(Ph)(H)}]”
5, which upon formation, undergo aldol-type reactions with unreacted substrate to give the observed products. NMR spectroscopic studies of the reaction mixtures prior to aqueous work-up did not show any evidence of 5 or the expected gallium alkoxide conjugate bases of 1a–3a. Instead, only 4 and the quenched products were present. This suggests that product quenching via proton abstraction from the toluene reaction solvent or another reaction component readily occurs. The increasing yield of 4 with increasing size of R could, therefore, be explained by a competition between solvent quenching of 5 (to give 4) and its nucleophilic attack on the α-alkoxy ketone substrate. The latter process would, presumably, be slowed with increasing size of the alkoxy substituent, thus favouring the formation of 4. When the reactions were repeated utilising benzene as the solvent, the resultant yields and diastereoselectivities were nearly identical to the toluene reactions. Intriguingly, when hexane was used as a solvent no reactions occurred. One explanation for this is that arene solvents are required to partly solubilise the “GaI” reagent. Indeed, it is well known that Ga(I) salts engage in π-arene interactions in solution.11
The diastereoselectivity of these reactions is interesting, but even more so is the change in the diastereoisomer formed upon changing the size of the substrate alkoxy group. It cannot be sure why these differences occur but analogies can be drawn with the previously reported additions of α-stannyl esters12 or indium enolates13 towards α-alkoxy ketones which give syn-products (cf. formation of 1a) with a very high degree of diasteroselectivity. It was proposed that this selectivity arose from a steric control of the enolate attack imposed by chelation of the substrate to the tin or indium centre prior to addition. In contrast, additions of indium enolates to simple ketones were found to proceed diastereoselectively to give anti-products (cf. formation of 2a and 3a). Here, six-membered cyclic transition states were used to explain the selectivity of the reactions. In our systems, it is feasible that the reaction of benzoin methyl ether with “GaI” could proceed via a chelated cyclic transition state (Scheme 1) which when quenched would lead to the observed diastereoisomer, 1a. It is possible that the bulkier alkoxy substituents of 2 and 3 preclude chelated transition states in their reactions with “GaI” and instead six-membered transition states, akin to those of indium enolate additions to simple ketones, lead to 2a and 3a upon quenching. If this is the case, the stereo-control of these reactions is dominated by the electrophillic substrate rather than the enolate. It is possible that the cyclic transition state that gave 2a and 3a is stabilised by chelation of its alkoxy and ketone O-centres to gallium salts (e.g. GaI3 from the disproportionation of “GaI”6a) which are likely to be present in the reaction mixture.
 | ||
| Scheme 1 Proposed mechanisms for the formation of 1a–3a. (i) “GaI”, toluene; (ii) quench. | ||
As the formulation of “GaI” is not definitely known, but is believed to include the mixed oxidation salt, GaI2[GaII2I6], it cannot be sure if the active component of the reagent is the Ga(I) cation, the Ga(II) anion, or both. In addition, “GaI” is well known to disproportionate to Ga metal and GaI3 in the presence of Lewis base donor sites,6a as are present in 1–3. Therefore, 1 was reacted with several Ga(II) and Ga(III) halides, and gallium metal under identical conditions to its reaction with “GaI” for purposes of comparison. The outcomes of these reactions are highlighted in Table 2 and show that no reaction occurs with gallium metal or gallium(III) halides, suggesting the formation of 1a involves a reduction process. This is perhaps confirmed by the reaction with the gallium(II) halide, a milder reducing agent than “GaI”, which only affords a very low yield of 1a. Similarly, no reaction occurred between 1 and InI, even under reflux conditions, which is consistent with our previous studies that point towards this being a milder reductant than “GaI”.8,9 Furthermore, when 1 was reacted with only two equivalents of “GaI”, a reduced yield of 1a was obtained and much of 1 remained unreacted. If the proposed mixed oxidation state formulation of “GaI” is correct, these results suggest that its Ga(I) cations are the active components of the salt. Finally, all attempts at in situ cross condensation reactions, for example between 1 and 2 or between 1 and ketones, were not successful and yielded only 1 and 4, or intractable mixtures of products.






Compounds 1a–3a were characterised by X-ray crystallography and the molecular structures of one enantiomer of each are depicted in Fig. 1. These were used to assign the stereochemistry of the products. To confirm that the crystals chosen for the X-ray experiments represented the bulk materials, their 1H NMR spectra were obtained and these were found to be identical to the total crystallised product. Each compound exhibits hydrogen bonding interactions between its ketone and alcohol functionalities, though the differences in the stereochemistries of the products mean that in 1a these interactions are intermolecular, leading to 1-dimensional hydrogen bonded polymers, whereas in 2a and 3a they are intramolecular.
 | ||
| Fig. 1 Molecular structures of (a) 1a, (b) 2a and (c) 3a (25% thermal ellipsoids are shown). | ||
(ii) Reactivity of “GaI” towards α-halo ketones
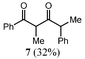
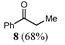
Considering that halides are better leaving groups than alkoxy substituents, it was thought that the reaction of α-halo ketones with “GaI” might lead to γ-halo β-hydroxy ketones (cf. the reactions that gave 1a–3a). Saying this, reactions of α-halo ketones with reducing agents such as SmI2 are known to facilely effect halide elimination and ketone formation.10 In the case of the reaction of the α-bromo ketone, 6, with “GaI”, this did occur to give the ketone, 8, as the major product, though a significant amount of the known diketone, 7, was also formed as a mixture of diastereoisomers (Table 3). We believe the mechanism of formation of 7 is similar to that reported for its synthesis from the reaction of 6 with MeMgBr.14 That is, the reaction proceeds through a dehalogenation step, to yield a gallium enolate which then attacks a second molecule of 6, leading to bromide elimination and a concomitant 1,2-phenyl migration yielding 7 (Scheme 2). This phenyl migration helps to explain the lack of diastereoselectivity in the reaction.| Substrate | Reagents | Products (yield) | |
|---|---|---|---|

|
4 ‘GaI’ |
|
|
| GaCl3 |

|

|
|
| 4 Ga2Cl4(dioxane)2 |

|

|
|

|
4 ‘GaI’ |

|

|
| GaCl3 |

|
— | |
| AlCl3 |

|

|
|
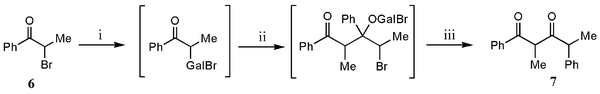 | ||
| Scheme 2 Proposed mechanism for the formation of 7. (i) “GaI”, toluene; (ii) 6; (iii) −GaIBr2, 1,2-phenyl migration. | ||
An 1H NMR spectroscopic analysis of the reaction mixture prior to aqueous work-up revealed the presence of both 7 and 8, suggesting that the intermediate gallium enolate readily abstracts a proton from the toluene solvent. This process would therefore be in competition with the aldol condensation that ultimately gives 7, thus leading to the observed mixture of 7 and 8. The apparent facile solvent quenching of the intermediate gallium enolate contrasts with work involving the reaction of GaMenI3−n with 6 which led to room temperature stable gallium enolates that underwent successful aldol reactions with carbonyl compounds and imines.15
In order to ascertain if the Ga(I) component of “GaI” plays the active role in the formation of 7, compound 6 was reacted with Ga(II) and Ga(III) halides under identical conditions. In these cases GaCl3 was largely unreactive, whilst treatment of 6 with Ga2Cl4(dioxane)2 almost exclusively gave the ketone, 8 (Table 3).
As the chloride group is a poorer leaving group than bromide, but a better leaving group than alkoxides, the reaction of an α-chloro ketone, 9, with four equivalents of “GaI” in toluene was carried out for purposes of comparison. This led to a completely different outcome to the reactions with 1–3 and 7, namely para-alkylation of the toluene solvent to give a low to moderate yield of the known ketone, 10.16 The previously observed quenched enolate, 4, was formed as the major product. The formation of 4 is not surprising in light of the fact that 9 is known to form enolates with other reducing agents, e.g. barium metal,17 but the absence of any coupled products (cf. 7) is unusual. More surprising is the apparent role of “GaI”, or one of its disproportionation products, as a Friedel–Crafts alkylation reagent in the formation of 10, which was not observed in any previous reaction. Evidence for this proposal comes from the results of the reactions of 9 with the classical Friedel–Crafts alkylation reagents, AlCl3 or GaCl3, which gave high or quantitative yields, respectively, of 10. Of course, no evidence for 4 was seen in these reactions due to the non-reducing nature of the salts involved. The X-ray crystal structure of 10 was determined and its ORTEP diagram is included in the supplementary material.
(iii) Reactivity of “GaI” towards other α-functionalised ketones
Low oxidation state metal complexes are well known to reduce 1,2-diketones to give chelated enediolate complexes.18 To examine the applicability of “GaI” to such reactions, it was treated with benzil, 11, in toluene. After aqueous work-up, benzoin, 12, was recovered in near quantitative yield (Scheme 3). Despite the reactivity of α-alkoxy ketones towards “GaI”, 12 was found to be completely unreactive to this reagent in toluene. This seems unusual, given the strongly reducing nature of “GaI” which one might think would lend it to the formation of 4, or dihydrobenzoin, PhC(OH)C(OH)Ph, in this reaction.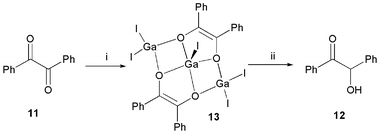 | ||
| Scheme 3 (i) “GaI”, toluene; (ii) quench. | ||
In order to shed light on the nature of the inorganic intermediate in the reaction that gave 12, the reaction was worked up prior to aqueous quench, affording a high yield of the novel trimetallic, bis(enediolato) complex, 13. Presumably, this forms by a combination of diketone reduction and “GaI” disproportionation processes. The molecular structure of 13 is depicted in Fig. 2. Surprisingly, this represents the first structural characterisation of a gallium enediolato complex. It possess three Ga(III) centres, two of which, Ga(2) and Ga(3), have distorted tetrahedral geometries and bridge an O-centre from each diolate ligand. The other gallium, Ga(1), has a distorted square based pyramidal geometry and is coordinated to all four O-centres. Although there are differences in the Ga–O and Ga–I bond lengths within the structure, all lie within the normal ranges19 and are unremarkable.
 | ||
| Fig. 2 Molecular structure of 13 (25% thermal ellipsoids are shown). Selected bond lengths (Å) and angles (°): I(1)–Ga(1) 2.4465(9), Ga(1)–O(3) 1.930(4), Ga(1)–O(2) 1.936(4), Ga(1)–O(1) 1.984(3), Ga(1)–O(4) 1.998(3), O(1)–C(1) 1.406(6), O(1)–Ga(3) 1.930(4), I(2)–Ga(2) 2.4618(10), Ga(2)–O(2) 1.913(4), Ga(2)–O(4) 1.943(3), Ga(2)–I(3) 2.4710(9), Ga(3)–O(3) 1.940(3), Ga(3)–I(4) 2.4617(8), Ga(3)–I(5) 2.4686(8), O(3)–Ga(1)–O(2) 123.00(16), O(3)–Ga(1)–O(1) 78.07(15), O(2)–Ga(1)–O(1) 81.27(15), O(3)–Ga(1)–O(4) 82.40(14), O(2)–Ga(1)–O(4) 78.84(14), O(1)–Ga(1)–O(4) 137.98(15), O(3)–Ga(1)–I(1) 123.29(11), O(2)–Ga(1)–I(1) 113.71(12), O(1)–Ga(1)–I(1) 112.27(11), O(4)–Ga(1)–I(1) 109.59(11), O(2)–Ga(2)–O(4) 80.78(15), O(2)–Ga(2)–I(2) 108.59(12), O(4)–Ga(2)–I(2) 110.09(11), O(2)–Ga(2)–I(3) 116.27(12), O(4)–Ga(2)–I(3) 112.41(11), I(2)–Ga(2)–I(3) 121.45(4), O(1)–Ga(3)–O(3) 79.15(15), O(1)–Ga(3)–I(4) 112.23(11), O(3)–Ga(3)–I(4) 113.93(11), O(1)–Ga(3)–I(5) 110.05(11), O(3)–Ga(3)–I(5) 111.18(11), I(4)–Ga(3)–I(5) 122.21(4). | ||
In keeping with the theme of this study, the reduction of a series of α,β-unsaturated ketones and related compounds with “GaI” was attempted. In all cases, no reduction products were returned, but in reactions with esters, deposition of gallium metal was observed. It is thought that this arises from a disproportionation of the gallium reagent to the metal and GaI3 upon coordination by the O-centres of the ester. Such disproportionation reactions have been previously reported for “GaI”.6a Indeed, in its reaction with trans-ethyl cinnamate in this study, a high yield of its GaI3 adduct, [GaI3{OC(OEt)C(H)C(H)Ph}] 14, was isolated and structurally characterised. Details of this structure can be found in the supplementary material.
Conclusions
In summary, the reactivity of “GaI” towards a range of α-functionalised ketones has been examined for the first time. Of most interest are the reactions of this reagent with α-alkoxy ketones which led to diastereoselective aldol coupling reactions and the formation of novel trifunctional products containing three contiguous stereocentres. The differences in the stereoselectivities of these reactions has been explained by invoking the involvement of two different transition states that depend upon the steric bulk of the alkoxy substituent. An aldol-type coupling reaction was also observed in the reaction of an α-bromo ketone with “GaI”, whilst in the reaction with a α-chloro ketone, the gallium reagent appeared to act as a Friedel–Crafts alkylation reagent. A selective, high yielding reduction of a 1,2-diketone to an α-hydroxy ketone has been achieved and the inorganic reaction intermediate structurally characterised. Throughout this work “GaI” has proved to be a stronger reducing agent than InI and has showed different chemistry to either Ga(II) or Ga(III) salts. The results of this study establish easy to prepare “GaI” as a potential specialist C–C bond forming reagent for the organic synthetic chemist. Work is ongoing in our laboratory to optimise the reported reactions and to explore new synthetic avenues for “GaI”, especially with regard to cross coupling reactions.Experimental section
General considerations
All manipulations were carried out using standard Schlenk and glove-box techniques under an atmosphere of high purity argon. The solvents toluene and hexane were distilled over potassium whilst diethyl ether was distilled over Na–K alloy. 1H and 13C NMR spectra were recorded on either a Bruker DXP400 spectrometer operating at 400.13 and 100 MHz, respectively or a JEOL Eclipse 300 spectrometer operating at 300.52 and 75.57 MHz, respectively. Spectra were referenced to either the 13C or residual 1H resonances of the solvent used. High resolution mass spectra were obtained from the EPSRC National Mass Spectrometric Service at Swansea University. IR spectra were recorded as Nujol mulls between NaCl plates. Melting points were determined in sealed glass capillaries under argon and are uncorrected. ‘GaI’,6 InI20 and Ga2Cl4(dioxane2)21 were synthesised by literature methods. All other reagents were obtained commercially and used as received.NB: an identical procedure was used for the reactions in Tables 2 and 3 that employed other inorganic reagents.
O); HRMS: (ES) calcd. for [M+ + Na]: 445.1774; found 445.1775.
O); HRMS: (ES) calcd for [MH+]: 437.2111; found 437.2110.
O); HRMS: (ES) calcd for [MH+]: 451.2268; found 451.2276.
O); HRMS: (EI) calcd for [C28H20O4Ga3127I5+]: 1261.4347; found 1261.4338; Anal. calcd for C28H20O4Ga3I5: C 26.60, H 1.59; Found: C 27.34, H 1.72.
X-Ray crystallography
Crystals of 1a–3a, 10, 13 and 14 suitable for X-ray structural determination were mounted in silicone oil. Crystallographic measurements were made using a Nonius Kappa CCD diffractometer. The structures were solved by direct methods and refined on F2 by full matrix least squares (SHELX-97)22 using all unique data. All non-hydrogen atoms are anisotropic with H-atoms included in calculated positions (riding model). Compounds 1a and 2a crystallised in chiral space groups but their X-ray analyses only established the relative stereochemistry of these “all light atom” structures. Compounds 3a and 10 crystallised in centrosymmetric space groups and are racemic. Crystal data, details of data collections and refinement are given in Table 4.| Compound | 1a | 2a | 3a | 10 | 13 | 14 |
|---|---|---|---|---|---|---|
| a CCDC 626737–626742. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b613669a | ||||||
| Empirical formula | C29H26O3 | C30H28O3 | C31H30O3 | C21H18O | C28H20Ga3I5O4 | C11H12GaI3O2 |
| FW | 422.50 | 436.52 | 450.55 | 286.35 | 1264.10 | 626.63 |
| Temp./K | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | C2 | P212121 | P21/n | P1 | P21/n | P21/n |
| a/Å | 32.142(6) | 6.0010(12) | 20.793(4) | 5.7983(12) | 9.0898(18) | 7.4458(15) |
| b/Å | 6.2276(12) | 18.790(4) | 6.1220(12) | 9.0444(18) | 19.066(4) | 18.197(4) |
| c/Å | 11.413(2) | 20.595(4) | 20.961(4) | 15.820(3) | 20.516(4) | 12.271(3) |
| α/° | 90 | 90 | 90 | 94.69(3) | 90 | 90 |
| β/° | 101.70(3) | 90 | 114.24(3) | 100.00(3) | 95.84(3) | 94.13(3) |
| γ /° | 90 | 90 | 90 | 106.81(3) | 90 | 90 |
| Vol./Å3 | 2237.1(8) | 2322.3(8) | 2433.0(8) | 774.4(3) | 3537.1(12) | 1658.3(6) |
| Z | 4 | 4 | 4 | 2 | 4 | 4 |
| Density (calc.)/Mg m−3) | 1.254 | 1.249 | 1.230 | 1.228 | 2.374 | 2.510 |
| μ(Mo-Kα) | ||||||
| /mm−1 | 0.080 | 0.079 | 0.078 | 0.074 | 6.663 | 7.233 |
| F(000) | 896 | 928 | 960 | 304 | 2312 | 1136 |
| No. of reflections collected | 5064 | 13489 | 7991 | 4993 | 15044 | 4253 |
| No. of independent reflns | 3292 | 4205 | 4444 | 2689 | 7690 | 2849 |
| R int | 0.1064 | 0.0683 | 0.0710 | 0.0381 | 0.0420 | 0.0499 |
| Final R indices [I > 2σ(I)] | R1 = 0.0876 | R1 = 0.0500 | R1 = 0.0554 | R1 = 0.0594 | R1 = 0.0399 | R1 = 0.0533 |
| wR2 = 0.1387 | wR2 = 0.0901 | wR2 = 0.1081 | wR2 = 0.1280 | wR2 = 0.0878 | wR2 = 0.1217 | |
Acknowledgements
We thank the EPSRC (partial studentships for S. P. G. and R. P. R.) for financial support. The EPSRC Mass Spectrometry Service at Swansea University is also thanked. Dr. Mark Bagley (Cardiff University) is thanked for helpful discussions.References
- See for example (a) T.-P. Loh and G.-L. Chua, Chem. Commun., 2006, 2739 RSC; (b) V. Nair, S. Ros, C. N. Jayan and B. S. Pillai, Tetrahedron, 2004, 60, 1959 CrossRef CAS; (c) M. R. Pitts, J. R. Harrison and C. J. Moody, J. Chem. Soc., Perkin Trans. 1, 2001, 955 RSC; (d) K. K. Chauhan and C. G. Frost, J. Chem. Soc., Perkin Trans. 1, 2000, 3015 RSC; (e) B. C. Ranu, Eur. J. Org. Chem., 2000, 2347 CrossRef CAS; (f) C.-J. Li and T.-H. Chan, Tetrahedron, 1999, 55, 11149 CrossRef CAS; (g) P. Cintas, Synlett, 1995, 1087 CrossRef CAS and references therein.
- “Main Group Metals in Organic Synthesis”, ed. H. Yamamota and K. Oshima, Wiley-VCH, Weinheim, 2004 Search PubMed.
- See for example (a) R. Amemiya and R. Yamaguchi, Eur. J. Org. Chem., 2005, 24, 5145 CrossRef; (b) R. M. Kellog and B. V. Syncom, Chemtracts, 2003, 16, 79 and references therein.
- (a) Y. Hashimoto, K. Hirata, H. Kagoshima, N. Kihara, M. Hasegawa and K. Saigo, Tetrahedron, 1993, 49, 5969 CrossRef CAS; (b) Y. Hashimoto, K. Hirata, N. Kihara, M. Hasegawa and K. Saigo, Tetrahedron Lett., 1992, 33, 6351 CrossRef CAS.
- R. J. Baker and C. Jones, Coord. Chem. Rev., 2005, 149, 1857 CrossRef and references therein.
- (a) R. J. Baker and C. Jones, Dalton Trans., 2005, 1341 RSC; (b) M. L. H. Green, P. Mountford, G. J. Smout and S. R. Peel, Polyhedron, 1990, 9, 2763 CrossRef CAS and references therein.
- S. Coban, Diplomarbeit, Universität Karlsruhe, 1999.
- R. J. Baker and C. Jones, Chem. Commun., 2003, 390 RSC.
- R. J. Baker, C. Jones, M. Kloth and D. P. Mills, New J. Chem., 2004, 28, 207 RSC.
- G. A. Molander and G. Hahn, J. Org. Chem., 1986, 51, 1135 CrossRef CAS.
- Chemistry of Aluminium Gallium Indium and Thallium, ed. A.J. Downs, Blackie Academic Press, Glasgow, 1993 Search PubMed.
- M. Yasuda, K. Okamoto, T. Sako and A. Baba, Chem. Commun., 2001, 157 RSC.
- (a) S. A. Babu, M. Yasuda, I. Shibata and A. Baba, J. Org. Chem., 2005, 70, 10408 CrossRef CAS; (b) S. A. Babu, M. Yasuda, Y. Okabe, I. Shibata and A. Baba, Org. Lett., 2006, 14, 3029 CrossRef.
- R. Nouri-Bimorghi, Bull. Soc. Chim. Fr., 1969, 8, 2812.
- Y. Han and Y.-Z. Huang, Tetrahedron Lett., 1998, 39, 7751 CrossRef CAS.
- L. Zhou and Y. Zhang, Tetrahedron, 2000, 56, 2953 CrossRef CAS.
- A. Yanagisawa, H. Takahashi and T. Arai, Chem. Commun., 2004, 580 RSC.
- See, for example: (a) L.-C. Song, P.-C. Liu, C. Han and Q.-M. Hu, J. Organomet. Chem., 2002, 648, 119 CrossRef CAS; (b) C. S. Weinert, A. E. Fenwick, P. E. Fanwick and I. P. Rothwell, Dalton Trans., 2003, 532 RSC; (c) J. Barrau, G. Rima and T. E. Amraoui, Organometallics, 1998, 17, 607 CrossRef CAS.
- As determined by a survey of the Cambridge Crystallographic Database, August, 2006.
- Synthetic Methods of Organometallic and Inorganic Chemistry, ed. W. A. Herrmann and G. Brauer, Thieme, Stuttgart, 1996, vol. 2, pp. 118 Search PubMed.
- P. Wei, X. Li and G. H. Robinson, Chem. Commun., 1999, 1287 RSC.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEP diagrams for 10 and 14. See DOI: 10.1039/b613669a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2007 |