DOI:
10.1039/B608680B
(Paper)
New J. Chem., 2007,
31, 135-143
Influence of ligand backbone flexibility in group 4 metal complexes of tetradentate mixed tertiary amine/alkoxide ligands†
Received
(in Durham, UK)
19th June 2006
, Accepted 9th November 2006
First published on 6th December 2006
Abstract
Simple epoxide ring opening chemistry using the cyclic secondary amine 1,4-diazacycloheptane or the related linear species N,N′-dimethylethylenediamine, and racemic (±)−3,3-dimethyl-1,2-epoxybutane gives access to the pendant alcohol functionalised ditertiary amine pro-ligands [HOCH(tBu)CH2N(R)CH2]2 (H2L1: R2 = CH2CH2CH2; H2L2: R2 = Me2). The contrasting reactions of H2L1 and H2L2 towards homoleptic group 4 alkoxides highlight the crucial role of ligand backbone flexibility in complex formation. Thus, the chemistry of the more conformationally rigid system (L1)2− appears to be constrained by the cyclic ligand core, such that it adopts a bridging (μ2:η2,η2) mode of coordination towards Ti(IV), leading to dinuclear metal systems [e.g.L1Ti2(OiPr)6]. By contrast, the more flexible linear system (L2)2− binds to both Ti(IV) and Zr(IV) in a chelating fashion leading, for example, to the synthesis of the C2 symmetric mononuclear complex rac-L2Ti(OiPr)2. Thus, a simple synthesis of diastereomerically pure, C2 symmetric, geometrically cis octahedral Ti(IV) complexes from racemic precursors is presented.
Introduction
The design and synthesis of ancillary ligand frameworks offering an alternative to cyclopentadienyl-based systems in early transition metal olefin polymerisation catalysts remains an area of intense research activity.1 Within this field, ligands featuring anionic oxygen or nitrogen donors (e.g. alkoxides/aryloxides, amides or imides) are particularly attractive targets given their π-donor capabilities and the strong electrostatic component to the metal–ligand interaction typically found with electropositive metals. For anionic oxygen ligands, RO−, the avoidance of secondary bridging interactions has led to the exploitation of a number of strategies in complex synthesis, e.g. the incorporation of steric bulk, chelating ligand frameworks and/or additional neutral donor atoms.2 Hence, for example chelating, sterically encumbered or heteroatom-functionalized bis(aryloxide) complexes of titanium and zirconium have been shown to be highly active olefin polymerisation catalysts,3–5 and alkoxide systems bearing ancillary N-donors have also been the subject of a number of recent studies.6
The use of metal complexes in asymmetric transformations, e.g. C2 symmetric (or pseudo-C2 symmetric) species in the isotactic polymerisation of propylene, has led to the development of a number of strategies for the synthesis of such complexes. Tetradentate bis(aryloxide) ligands featuring a linear array of donor atoms (Scheme 1) have featured prominently among these, with examples of both pre-constructed C2 symmetric pro-ligands4b,f,g,5m and coordination induced C2 symmetry having been reported.4d,k,u,5p,6b,7 Within this area we have been interested in developing synthetic routes to sterically encumbered bis(tertiary amine) bis(alkoxide) ligand frameworks, through the ring opening of (±)-3,3-dimethyl-1,2-epoxybutane (Scheme 2). It was envisaged that the use of racemic precursors and the separation of diastereomers by simple crystallization at either the pro-ligand or complex stage would provide a convenient route to C2 symmetric species.
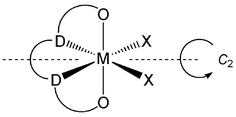 |
| | Scheme 1 Target C2 symmetric, geometrically cis, octahedral complexes featuring tetradentate bis(donor) bis(aryl/alkoxide) ligands. | |
![Syntheses of ligand precursors H2L1 and H2L2. Reagents and conditions: excess (±)-3,3-dimethyl-1,2-epoxybutane, acetonitrile, sealed tube, 100 °C, 72 h, yield ca. 40% for H2L1 [obtained as the rac (R,R/S,S) diastereomers], 59% for H2L2 [obtained as a mixture of the rac (R,R/S,S) and meso (R,S) diastereomers].](/image/article/2007/NJ/b608680b/b608680b-s2.gif) |
| | Scheme 2 Syntheses of ligand precursors H2L1 and H2L2. Reagents and conditions: excess (±)-3,3-dimethyl-1,2-epoxybutane, acetonitrile, sealed tube, 100 °C, 72 h, yield ca. 40% for H2L1 [obtained as the rac (R,R/S,S) diastereomers], 59% for H2L2 [obtained as a mixture of the rac (R,R/S,S) and meso (R,S) diastereomers]. | |
Experimental
(i) General considerations
Unless otherwise stated, all manipulations were carried out under a nitrogen or argon atmosphere using standard Schlenk line or dry-box techniques. Solvents were pre-dried over sodium wire (hexanes, toluene) or molecular sieves (acetonitrile) and purged with nitrogen prior to distillation from the appropriate drying agent (hexanes: potassium, toluene: sodium, acetonitrile: calcium hydride). Benzene-d6 and chloroform-d (both Goss) were degassed and dried over potassium (benzene-d6) or molecular sieves (chloroform-d) prior to use. Ligand precursors [1,4-diazacycloheptane (Lancaster), 1,4-diazacyclohexane (Avocado), N,N′-dimethylethylenediamine (Aldrich) and (±)-3,3-dimethyl-1,2-epoxybutane (Lancaster)] and metal reagents [Ti(OiPr)4 (Lancaster), Ti(OEt)4 and Zr(OnPr)4 (all Aldrich)] were used as received. NMR spectra were measured on a Bruker AM-400 or JEOL 300 Eclipse Plus FT-NMR spectrometer. Residual signals of solvent were used as reference for 1H and 13C NMR. Infrared spectra were measured for each compound pressed into a disk with an excess of dried KBr or as a solution in an appropriate solvent on a Nicolet 500 FT-IR spectrometer. Mass spectra were measured by the EPSRC National Mass Spectrometry Service Centre, University of Wales, Swansea. Perfluorotributylamine was used as a standard for high resolution EI mass spectra. Elemental microanalysis was performed by Warwick Analytical Services or by the departmental service. Abbreviations: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, m = multiplet.
(ii) Ligand syntheses
N,N′-Bis(2-hydroxy-3,3-dimethylbutyl)-1,4-diazacycloheptane (H2L1).
To a solution of 1,4-diazacycloheptane (0.90 g, 8.99 mmol) in acetonitrile (10 ml), contained in a pressure tube, was added excess (±)-3,3-dimethyl-1,2-epoxybutane (2.69 g, 26.86 mmol). The reaction mixture was stirred at 100 °C for 24 h, and the white crystals which formed on cooling of the reaction mixture were filtered off and washed with cold acetonitrile. Although single crystals suitable for X-ray diffraction could not be obtained, the 1H and 13C NMR data for crystalline samples re-dissolved in chloroform-d imply that only one set of diastereomers is present; the crystal structures of nickel(II) and titanium(IV) complexes obtained by further reaction chemistry indicate that this is the rac (R,R/S,S) pair. Yield: 1.15 g, 43%. 1H NMR (CDCl3, 25 °C): δ 0.85 (s, 18 H, tBu), 1.85 (m, 2H, NCH2CH2CH2N), 2.20 (m, 2H, C(H)(H)CHOH), 2.50 (m, 2H, C(H)(H)CHOH), 2.55 (m, 4H, NCH2), 2.82 (m, 4H, NCH2), 3.22 (m, 2H, CHOH), 3.82 (br s, 2H, OH). 13C NMR (CDCl3, 25 °C): δ 25.7 (CH3 of tBu), 28.2 (NCH2CH2CH2N), 33.2 (tBu quaternary), 54.4 (NCH2), 56.2 (NCH2), 59.5 (CH2CHOH), 73.7 (CHOH). IR (KBr disk, cm−1) 3417, 2955, 2855, 1653, 1467, 1410, 1362, 1244, 1154, 1088, 1013, 944, 863, 821. Mass spectrum (APCI): 301.6 (M + H)+. Elemental analysis. Calc. for C17H36N2O2: C, 67.95; H, 12.08; N, 9.32. Found: C, 68.31; H, 12.55; N, 8.99%. In an analogous manner, the corresponding reaction using 1,4-diazacyclohexane generates crystalline samples of N,N′-bis(2-hydroxy-3,3-dimethylbutyl)-1,4-diazacyclohexane which can be shown by a combination of NMR and X-ray crystallographic studies to contain solely the meso (R,S) isomer.8
N,N′-Bis(2-hydroxy-3,3-dimethylbutyl)dimethylethylenediamine (H2L2).
H2L2 was synthesized in an analogous manner to H2L1, using N,N′-dimethylethylenediamine (0.50 g, 5.67 mmol) and isolated after removal of volatiles from the reaction mixture in vacuo as a clear oil containing a mixture of rac (R,R/S,S) and meso (R,S) diastereoisomers at room temperature (waxy solid at −25 °C). Yield: 0.63 g, 59%. 1H NMR (CDCl3, 25 °C): δ 0.85 (s, 36 H, coincident tBu groups of both pairs of diastereomers), 2.23, 2.26 (s, each 6H, NCH3), 2.31–2.68 (br overlapping m, 16H, overlapping NCH2 groups of both diastereomers), 3.24, 3.27 (m, each 2H, CHOH), 4.31, 4.53 (br s, each 2H, OH). 13C NMR (CDCl3, 25 °C): δ 25.87, 25.89 (CH3 of tBu), 33.4 (coincident tBu quaternary carbons of both pairs of diastereomers), 42.5, 43.5 (NCH3), 54.7, 55.8 (NCH2), 58.6, 59.2 (CH2CHOH), 74.6, 74.7 (CHOH). IR (KBr disk, cm−1) 2957, 1463, 1363, 1217, 1090, 1017, 937. Mass spectrum (APCI): 289.1 (M + H)+. Exact mass. Calc. for C16H36N2O2: 289.2855. Found: 289.2859. Accurate elemental analysis proved impossible for this oil.
(iii) Complex syntheses
L1Ti2(OiPr)6.
Neat titanium tetrakis(isopropoxide) (0.50 ml, 1.76 mmol) was added to a H2L1 (0.50 g, 1.66 mmol) and the reaction mixture stirred until it became solid (ca. 4 h). Isopropanol formed during the reaction was then removed in vacuo and the resulting solid redissolved in dry hexanes. Colourless crystals suitable for X-ray diffraction were grown by cooling the hexanes solution to −30 °C. Variation in reaction conditions (stoichiometry of reaction, order of reagent addition, temperature) did not result in the isolation of alternative products containing a different Ti : L1 ratio. The yield of L1Ti2(OiPr)6 was optimised by consideration of the 2 : 1 Ti : L1 ratio to 64% (ca. 0.75 g scale). 1H NMR (C6D6, 25 °C): δ 1.00 (s, 18H, tBu), 1.37 (d, J = 6.0 Hz, 36H, CH3 of iPr), 1.85 (m, 2H, NCH2CH2CH2N), 2.55 (m, 2H, CH(H)CHO), 2.84 (m, 2H, CH(H)CHO), 3.50 (br overlapping m, 8H, NCH2 of diazacycloheptane), 4.08 (m, 2H, C(H)O), 4.92 (septet, J = 6.0 Hz, 6H, CH of iPr). 13C NMR: δ 26.3 (CH3 of tBu), 27.8 (CH3 of iPr), 31.1 (NCH2CH2CH2N), 35.6 (tBu quaternary), 58.1 (NCH2CH2CH2N), 60.1 (NCH2CH2N), 62.3 (CH2C(H)O), 77.4 (CH of iPr), 83.1 (CHO). Mass spectrum (FAB): 747.3 (M − H)+. Elemental analysis. Calc. for C35H76N2O8Ti2: C, 56.15; H, 10.23; N, 3.74. Found: C, 55.81; H, 10.01; N, 3.89%.
[L1Ti(OEt)]2(μ2-O).
[L1Ti(OEt)]2(μ2-O) was prepared from titanium tetrakis(ethoxide) (0.40 ml, 1.75 mmol) and H2L1 (0.50 g, 1.66 mmol) using the method detailed above for L1Ti2(OiPr)6. The product was isolated in very low yield (<5%) as colourless crystals obtained from hexanes solution at −30 °C. Complete characterization was frustrated in this case by the small amount of compound obtained. 1H NMR (C6D6, 25 °C): δ 0.99 (s, 36H, tBu), 1.21 (t, J = 8.0 Hz, 6H, OCH2CH3), 1.88 (m, 4H, NCH2CH2CH2N), 2.68 (br m, 4H, CH(H)CHO), 2.78 (m, 4H, CH(H)CHOH), 3.20–3.68 (br overlapping m, 16H, NCH2 of diazacycloheptane), 3.91 (q, J = 8.0 Hz, 4H, OCH2CH3), 4.15 (m, 4H, CHO). Mass spectrum (FAB): 799.6 (M − H)+.
L2Ti(OiPr)2.
L2Ti(OiPr)2 was prepared from titanium tetrakis(isopropoxide) (0.50 ml, 1.76 mmol) and H2L2 (0.50 g, 1.74 mmol) using the method detailed above for L1Ti2(OiPr)6, and isolated as colourless crystals after two recrystallizations from concentrated hexanes solution (yield: 44%, ca. 1 g scale). Although the oily ligand precursor H2L2 was used as a mixture of rac (R,R/S,S) and meso (R,S) isomers, X-ray diffraction analysis of the crystalline titanium complex reveals it has approximate (non-crystallographically imposed) C2 symmetry and contains the rac (R,R/S,S) isomers of the ligand. The oily hexanes-soluble residue remaining after recrystallization can be shown by 1H and 13C NMR to be the C1 symmetric complex derived from the meso (R,S) ligand precursor. Attempts to obtain this second isomer in pure form for comparative structural and catalytic studies were frustrated by its oily nature and the difficulty in removing the last traces of the C2 isomer. Characterizing data for crystalline product (R,R/S,S): 1H NMR (C6D6, 25 °C): δ 1.15 (s, 18H, tBu), 1.32 (d, J = 6.3 Hz, 6H, CH3 of iPr), 1.38 (d, J = 9.0 Hz, 2H, NCH2CH2N), 1.50 (d, J = 6.0 Hz, 6H, CH3 of iPr), 2.14 (dd, J = 10.7, 4.1 Hz, 2H, NCH2CHO), 2.22 (s, 6H, NCH3), 2.74 (d, J = 9.0 Hz, 2H, NCH2CH2N), 3.09 (virtual t, J = 11.0 Hz, 2H, CH2CHO), 3.84 (dd, J = 10.6, 4.1 Hz, 2H, NCH2CHO), 5.14 (sept, J = 6.0 Hz, 2H, CH of iPr). 13C NMR (C6D6, 25 °C): δ 26.0, 26.7 (CH3 of iPr), 26.4 (CH3 of tBu), 36.5 (tBu quaternary), 45.3 (NCH3), 51.9 (NCH2 backbone), 62.8 (NCH2 ligand arm), 74.6 (CH of iPr), 84.2 (CHO). Mass spectrum (EI): 452.4 M+. Exact mass. Calc. for TiC22H48N2O4 452.3088: Found: 452.3079. Elemental analysis. Calc. for TiC22H48N2O4: C, 58.40; H, 10.69; N, 6.19. Found: C, 58.01; H, 10.14; N, 6.24%. 1H and 13C NMR data for oily product (R,S): 1H NMR (C6D6, 25 °C): δ 0.93 (s, 9H, tBu), 1.03 (s, 9H, tBu), 1.29 (d, J = 6.0 Hz, 3H, CH3 of iPr), 1.33 (d, J = 6.0 Hz, 3H, CH3 of iPr), 1.39 (d, J = 3.0 Hz, 6H, CH3 of iPr), 1.44 (d, J = 3.0 Hz, 6H, CH3 of iPr), 1.71 (dd, J = 12.4, 4.1 Hz, 1H, NCH2CHO), 1.85 (dd, J = 14.0, 4.0 Hz, 1H, NCH2CHO), 2.16 (m, 2H, NCH2CH2N), 2.41 (s, 3H, NCH3), 2.42 (s, 3H, NCH3), 2.75 (m, 2H, NCH2CH2N), 3.06 (virtual t, J = 11.0 Hz, 1H, CH2CHO), 3.13 (virtual t, J = 11.0 Hz, 1H, CH2CHO), 3.79 (dd, J = 10.8, 4.4 Hz, 1H, NCH2CHO), 4.33 (dd, J = 9.8, 5.2 Hz, 1H, NCH2CHO), 4.96 (sept, J = 6.0 Hz, 2H, coincident CH of iPr). 13C NMR (C6D6, 25 °C): δ 26.0, 26.2 (CH3 of tBu), 25.9, 26.2, 26.6, 26.9 (CH3 of iPr), 34.6 (coincident tBu quaternary carbons), 47.9, 50.2 (NCH3), 56.7, 59.2 (NCH2 backbone), 59.4, 66.0 (NCH2 ligand arm), 74.7, 74.8 (CH of iPr), 84.8, 85.1 (CHO).
(L2)2Zr.
(L2)2Zr was prepared from zirconium tetrakis(propoxide) (0.78 ml of a 70% wt solution in propanol, 1.74 mmol) and H2L2 (0.50 g, 1.74 mmol) using the method detailed above for L1Ti2(OiPr)6, and isolated as colourless crystals from a concentrated hexanes solution. In contrast to the crude reaction mixture, the crystalline product gives rise to relatively simple 1H and 13C NMR spectra indicating only two distinct tBu and NMe resonances, with subsequent crystallographic analysis revealing both (L2)2− ligands to be of meso (R,S) stereochemistry. The yield was subsequently optimised by consideration of the 1 : 2 Zr : L2 ratio to 39% (ca. 1.00 g scale). 1H NMR (C6D6, 25 °C): δ 1.08 (s, 18H, tBu), 1.10 (s, 18H, tBu), 1.81 (m, 4H, NCH2CHO), 2.14 (s, 6H, NCH3), 2.34 (m, 4H, NCH2CH2N), 2.80 (m, 4H, NCH2CH2N), 2.91 (s, 6H, NCH3), 3.14 (t, J = 12.0 Hz, 2H, CH2CHO), 3.29 (t, J = 11.6 Hz, 2H, CH2CHO), 3.78 (dd, J = 2.6, 11.1 Hz, 2H, NCH2CHO), 4.09 (dd, J = 5.1, 11.4 Hz, 2H, NCH2CHO). 13C NMR (C6D6, 25 °C): δ 26.9, 27.2 (CH3 of tBu), 35.2, 35.3 (tBu quaternary), 43.4, 48.6 (NCH3), 57.2, 58.9 (NCH2CH2N), 60.4, 64.5 (NCH2), 78.9, 83.9 (CHO). Mass spectrum (FAB): 661.3 (M − H)+. Exact mass. Calc. for ZrC32H68N4O4: 661.4204. Found: 661.4187. Elemental analysis. Calc. for ZrC32H68N4O4: C, 57.87; H, 10.32; N, 8.44. Found: C, 57.41; H, 10.00; N, 8.59%.
(iv) Crystallographic method
Data for L1Ti2(OiPr)6, L12Ti2(OEt)2(μ-O), L2Ti(OiPr)2 and L22Zr were collected on an Bruker Nonius Kappa CCD diffractometer. Data collection and cell refinement were carried out using DENZO and COLLECT; structure solution and refinement used SHELXS-97 and SHELXL-97, respectively; absorption corrections were performed using SORTAV.9 Details of each data collection, structure solution and refinement can be found in Table 1. Relevant bond lengths and angles are included in the figure captions and complete details of each structure have been deposited with the CCDC (numbers as listed in Table 1). In addition, complete details for each structure have been included in the supporting information. The two OEt ligands in L12Ti2(OEt)2(μ-O) are disordered and were modeled over two orientations (70 : 30 and 53 : 47 occupancy factors) and refined isotropically. The structure was treated as a racemic twin with the BASF parameter refining to 0.4.
Table 1 Details of data collection, structure solution and refinement for L1Ti2(OiPr)6, L12Ti2(OEt)2(μ-O), L2Ti(OiPr)2 and L22Zr (refinement method: full-matrix least squares (F2))
| Compound |
L1Ti2(OiPr)6 |
L1
2Ti2(OEt)2(μ-O) |
L2Ti(OiPr)2 |
L2
2Zr |
| Empirical formula |
C35H76N2O8Ti2 |
C38H78N4O7Ti2 |
C22H48NO4Ti |
C32H68N4O4Zr |
|
M
r
|
748.78 |
798.84 |
452.52 |
664.12 |
|
T/K |
150(2) |
120(2) |
120(2) |
120(2) K |
| CCDC no. |
286 915 |
286 913 |
286 916 |
286 914 |
|
λ/Å |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| Crystal system |
Triclinic |
Orthorhombic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pca21 |
P |
P |
|
a/Å |
11.0190(5) |
19.7850(10) |
8.830(5) |
10.762(2) |
|
b/Å |
14.3144(6) |
11.7253(4) |
9.726(5) |
12.115(2) |
|
c/Å |
15.7604(8) |
19.6134(7) |
17.047(5) |
15.963(3) |
|
α/° |
70.240(3) |
90 |
93.801(5) |
76.68(3) |
|
β/° |
88.145(2) |
90 |
98.098(5) |
72.59(3) |
|
γ/° |
71.787(2) |
90 |
112.844(5) |
68.58(3) |
|
V/Å3 |
2214.83(18) |
4550.0(3) |
1323.9(11) |
1831.5(6) |
|
D
c/Mg m−3 |
1.123 |
1.166 |
1.135 |
1.204 |
|
Z
|
2 |
4 |
2 |
2 |
|
μ/mm−1 |
0.404 |
0.397 |
0.349 |
0.337 |
|
F(000) |
816 |
1736 |
496 |
720 |
| Crystal size/mm |
0.20 × 0.18 × 0.15 |
0.20 × 0.15 × 0.05 |
0.30 × 0.20 × 0.02 |
0.20 × 0.20 × 0.20 |
|
θ Range/° |
3.05–25.39 |
2.92–25.02 |
2.98–25.03 |
2.92–27.45 |
| Index ranges hkl |
−13 to 13, −17 to 17, −18 to 19 |
−23 to 18, −10 to 13, −23 to 21 |
−10 to 10, −11 to 11, −20 to 20 |
−13 to 13, −15 to 15, −10 to 20 |
| Reflections collected |
31 559 |
22 572 |
13 694 |
36 431 |
| Independent reflections (Rint) |
8020 (0.1319) |
7502 (0.0809) |
4583 (0.0501) |
8322 (0.0562) |
| Completeness to θmax (%) |
98.5 |
99.2 |
97.8 |
99.4 |
| Absorption correction |
SORTAV |
Semi-empirical from equivs |
Semi-empirical from equivs |
SORTAV |
| Max., min. transmission |
0.9419, 0.9236 |
0.9804, 0.9249 |
0.9930, 0.9025 |
0.9356, 0.8956 |
| Data/restraints/parameters |
8020/0/442 |
7502/9/471 |
4583/0/275 |
8322/0/407 |
| Goodness-of-fit on F2 |
1.002 |
1.008 |
1.030 |
1.032 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0684, wR2 = 0.1524 |
R1 = 0.0607, wR2 = 0.1248 |
R1 = 0.0416, wR2 = 0.0881 |
R1 = 0.0373, wR2 = 0.0851 |
|
R Indices (all data) |
R1 = 0.1451, wR2 = 0.1758 |
R1 = 0.1033, wR2 = 0.1410 |
R1 = 0.0538, wR2 = 0.0932 |
R1 = 0.0458, wR2 = 0.0886 |
| Δρmax, min/e Å−3 |
0.451, −0.427 |
0.618, −0.314 |
0.209, −0.366 |
1.117, −0.520 |
CCDC reference numbers 286913–286916.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b608680b
Results and discussion
(i) Ligand precursors
The ligand precursors H2L1 and H2L2 have been synthesised via the nucleophilic ring opening of racemic (±)-3,3-dimethyl-1,2-epoxybutane by the cyclic secondary amine 1,4-diazacycloheptane, or the related linear species N,N′-dimethylethylenediamine (to give H2L1 and H2L2, respectively; Scheme 2). Forcing conditions (excess epoxide, acetonitrile solution at 100 °C in a pressure tube) are required to drive the reaction to completion, the analogous chemistry in ethanol at room temperature leading to incomplete conversion. Similar chemistry can be also applied to 1,4-diazacyclohexane to yield the related compound N,N′-bis(2-hydroxy-3,3-dimethylbutyl)-1,4-diazacyclohexane.8 H2L1 (like its diaminocyclohexane analogue) is a white crystalline solid which has been characterised by multinuclear NMR and IR spectroscopies, mass spectrometry and elemental analysis. In both cases, the 1H and 13C NMR spectra (in chloroform-d) of crystalline ligand samples obtained by direct cooling of the acetonitrile reaction mixture, show single resonances for the tert-butyl methyl groups, indicating the presence of only one pair of diastereomers [i.e. either rac (R,R/S,S) or meso (R,S)]. In the case of N,N′-bis(2-hydroxy-3,3-dimethylbutyl)-1,4-diazacyclohexane, a crystallographic study reveals a centrosymmetric space group, in which each molecule lies on a centre of inversion coincident with the centre of the 1,4-diazacyclohexane ring. Consequently, this ligand necessarily features the meso (R,S) stereochemistry.8 Although crystalline H2L1 can also be obtained by direct cooling of the acetonitrile reaction mixture, the crystals so obtained are not suitable for X-ray diffraction. In this case, however, crystallographic studies (i) of alkoxo-titanium complexes formed by reaction of H2L1 with Ti(OR)4 (R = iPr, Et) (vide infra); and (ii) of the complex [trans-(H2L1)Ni(OH2)2]2+ isolated as the bis(perchlorate) salt from the subsequent reaction of crystalline H2L1 with Ni(ClO4)2·6H2O in ethanol,10 confirm that H2L1 crystallises from the reaction mixture as the rac (R,R/S,S) pair of diastereomers. Yields of crystalline rac-H2L1 and meso-N,N′-bis(2-hydroxy-3,3-dimethylbutyl)-1,4-diazacyclohexane are typically of the order of 40%, reflecting the fact that in each case the alternative pair of diastereomers remains in solution. In contrast, H2L2 can only be obtained as an oily liquid at room temperature (cooling to a waxy solid at −25 °C). 1H and 13C NMR spectra clearly indicate the presence of both pairs of diastereoisomers, the separation of which by fractional crystallisation proves impossible. Consequently, separation of diastereomeric species at the metal complex stage, making use of the differential solubilities of more tractable derivatives was thought to offer a more practical methodology (vide infra).
(ii) Complexes of group 4 metals
The coordination chemistries of ligands (L1)2− and (L2)2− with respect to group 4 metal centres have been investigated via the reactions of H2L1 and H2L2 with homoleptic titanium and zirconium tetrakis(alkoxide) precursors (Schemes 3 and 4).
 |
| | Scheme 3 Syntheses of dinuclear titanium complexes L1Ti2(OiPr)6 and L12Ti2(OEt)2(μ-O). Reagents and conditions: (i) Ti(OiPr)4 (2 equiv.), neat reagents, 20 °C, 4 h, 64%; (ii) Ti(OEt)4 (1.05 equiv.), neat reagents, adventitious water, 20 °C, 4 h, <5%. | |
 |
| | Scheme 4 Syntheses of mononuclear group 4 complexes L2Ti(OiPr)2 and L22Zr. Reagents and conditions: Ti(OiPr)4 or Zr(OnPr)4 (1 equiv.), neat reagents, 20 °C, 4 h, separation of isomers by recrystallization from hexanes, 44 and 39%, respectively (for crystalline products). | |
(a) Coordination chemistry of (L1)2−.
The reactions of H2L1 with titanium alkoxides lead to the formation of dinuclear complexes in which the tetradentate ligand bridges between two Ti(IV) centres. Thus, the reaction of rac-H2L1 with titanium tetrakis(isopropoxide) at room temperature in the absence of solvent gives a white crystalline solid, for which the integration of tBu and iPr 1H NMR signals (1 : 2) implies an L1 : OiPr ratio of 1 : 6. The formulation L1Ti2(OiPr)6 is given further credence by the results of mass spectrometry experiments and has been confirmed crystallographically (Fig. 1 and Table 1).
 |
| | Fig. 1 Structure of L1Ti2(OiPr)6; hydrogen atoms have been omitted for clarity and ORTEP ellipsoids set at the 30% probability level. Important bond lengths (Å) and angles (°): Ti(1)–O(1) 1.853(3), Ti(1)–O(2) 1.784(3), Ti(1)–O(3) 1.821(3), Ti(1)–O(4) 1.820(3), Ti(1)–N(1) 2.378(3), Ti(2)–N(2) 2.391(3); O(1)–Ti(1)–O(3) 119.3(1), O(1)–Ti(1)–O(4) 118.4(1); O(3)–Ti(1)–O(4) 115.9(1), O(2)–Ti(1)–N(1) 171.0(1), O(6)–Ti(2)–N(2) 170.38(12). | |
The X-ray crystal structure of L1Ti2(OiPr)6 shows that the complex contains two Ti(OiPr)3 fragments linked by a single bridging (L1)2− ligand which binds to each metal centre through one alkoxide and one tertiary amine donor. Each titanium centre is thus five coordinate, with three isopropoxide ligands being retained from the starting material. The geometry around each titanium atom appears to be a distorted trigonal bipyramid with the L1 amine donor and one isopropoxide ligand occupying the axial sites [O(2)–Ti(1)–N(1) 171.0(1)°, O(6)–Ti(2)–N(2) 170.4(1)°]. The L1-derived alkoxide donor and the two remaining isopropoxides occupy the three equatorial sites [O(1)–Ti(1)–O(3) 119.3(1)°, O(1)–Ti(1)–O(4) 118.4(1)°; O(3)–Ti(1)–O(4) 115.9(1)°]. In general terms, the Ti–O distances are within the bounds expected for five-coordinate titanium complexes of this type, with the Ti–N distance [2.385 Å (mean)] being, if anything, slightly longer than those found in comparable systems. Thus, for example, a useful comparison can be made with the corresponding bond lengths found in the complexes N[CH2(3,5-Me2C6H2)O]3TiOR [R = C6H3iPr2-2,6, d(Ti–O) = 1.833 Å (mean), d(Ti–N) = 2.305(2) Å; R = iPr, d(Ti–O) = 1.831 Å (mean), d(Ti–N) = 2.295(3) Å] and N[CH2(3,5-Me2C6H2)O]2(CH2CH2O)TiOR [R = C6H3iPr2-2,6, d(Ti–O) = 1.822 (mean), d(Ti–N) = 2.288(3) Å], each of which features an analogous (approximately trigonal bipyramidal) Ti(OR)4(NR3) unit, with the amine donor occupying one of the axial positions.11
In a bid to obtain a complex of the desired composition, i.e.L12Ti(OR)2, the reaction stoichiometry was varied (using a large excess of H2L1 under more forcing conditions); the same dinuclear complex was isolated. Reduction in the steric bulk of the alkoxide co-ligands was therefore investigated in order to probe whether this would allow for the coordination of more than one (L1)2− moiety. Reaction of H2L1 with titanium tetrakis(ethoxide) under similar conditions yields colourless crystals in low yield after recrystallisation from hexane. Integration of the tBu and Et 1H NMR signals implies an L1 : OEt ratio of 1 : 1, and the formulation L12Ti2(OEt)2(μ-O) demonstrated by mass spectrometry has been confirmed crystallographically (Fig. 2 and Table 1).
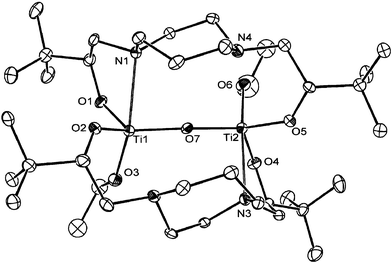 |
| | Fig. 2 Structure of L12Ti2(OEt)2(μ-O); hydrogen atoms have been omitted for clarity and ORTEP ellipsoids set at the 30% probability level. Important bond lengths (Å) and angles (°): Ti(1)–O(7) 1.826(4), Ti(2)–O(7) 1.818(4), Ti(1)–O(1) 1.860(4), Ti(1)–O(2) 1.860(4), Ti(1)–O(3) 1.835(3), Ti(2)–O(4) 1.852(3), Ti(2)–O(5) 1.840(4), Ti(2)–O(6) 1.827(4), Ti(1)–N(1) 2.506(4), Ti(1)–N(2) 2.757(4), Ti(2)–N(3) 2.443(4), Ti(2)–N(4) 2.962(4); O(6)–Ti(2)–N(3) 165.3(2), O(4)–Ti(2)–O(5) 109.4(2), O(4)–Ti(2)–O(7) 116.6(2), O(5)–Ti(2)–O(7) 125.0(2). | |
The crystal structure of L12Ti2(OEt)2(μ-O) confirms that a 1 : 1 ratio of (L1)2− to Ti(IV) can be achieved, albeit with the ligand still adopting a bridging, rather than chelating mode of binding. Noteworthy is the linking of the two titanium centres via a symmetrically bridging oxo ligand [Ti(2)–O(7) 1.818(4) Å, Ti(1)–O(7) 1.826(4) Å], which is presumably derived from conversion of titanium-bound OEt ligands to OH (by adventitious water) followed by a condensation step. The coordination sphere at each titanium also features one ethoxide ligand remaining from the starting material and two alkoxide linkages (one from each L1 ligand). Each metal centre is also engaged in two disparate Ti–N interactions [e.g. Ti(2)–N(3) 2.443(4), Ti(2)–N(4) 2.962(4) Å]. The shorter Ti–N distance is relatively long for a N→Ti donor/acceptor interaction featuring a Ti(OR)4 type Lewis acid,11 whereas the longer Ti–N distance falls outside the sum of conventional covalent radii for N and Ti.12 The geometry at each titanium centre can therefore probably best be considered as intermediate between trigonal bipyramidal and octahedral, being formally derived from a regular trigonal bipyramid by approach of the weakly bound sixth donor in the equatorial plane (Scheme 5). The axial positions are occupied by the more tightly bound amine donor and the ethoxide ligand [O(6)–Ti(2)–N(3) 165.3(2)°]. The two remaining L1 alkoxide donors and the bridging oxygen constitute the trigonal plane (sum of the O–Ti–O angles 351.0°) in which distortions from 120° angles presumably reflect, at least in part, the approach of the second, weakly-bound amine donor [O(4)–Ti(2)–O(5) 109.4(2)°; O(4)–Ti(2)–O(7) 116.6(2)°; O(5)–Ti(2)–O(7) 125.0(2)°].
 |
| | Scheme 5 Schematic representation of the coordination environment at titanium in L12Ti2(OEt)2(μ-O). | |
Preliminary results therefore imply that the relatively rigid ligand backbone in (L1)2− prevents it from binding in a chelating fashion to small transition metal centres such as Ti(IV). Furthermore, although the formation of a Ni(II) complex containing the diprotonated ligand H2L1 has demonstrated the possibility of chelation with larger metals, the ancillary water ligands in [(H2L1)Ni(OH2)2][ClO4]2 are coordinated in the undesirable trans orientation.10 The Ti(IV) and Zr(IV) coordination chemistries of the more flexible system (L2)2− were therefore targeted.
(b) Coordination chemistry of (L2)2−.
In the case of ligand (L2)2−, the precursor H2L2 is obtained as an inseparable mixture of rac and meso diastereomers and utilised as such for reactions with Ti(OiPr)4 and Zr(OnPr)4 (see Scheme 5). The reaction of H2L2 with titanium tetrakis(isopropoxide) gives two products, as determined from the 1H and 13C NMR spectra of the crude reaction mixture. Careful recrystallization from hexanes (twice) allows the isolation of a colourless crystalline solid, for which 1H and 13C NMR and mass spectrometry indicate a formulation as L2Ti(OiPr)2. The 13C NMR spectrum (see ESI†) shows only nine signals, indicating that in this complex the two halves of the ligand are symmetry related. This inference is confirmed by the results of a single-crystal X-ray diffraction study (Fig. 3 and Table 1) which confirms local (non-crystallographic) C2 symmetry and that the coordinated ligands are of rac stereochemistry (both R,R and S,S enantiomers being present in the crystal lattice). Overall, the titanium centre is six-coordinate (a distorted octahedron) and bound to a single L2 ligand, with two mutually cis isopropoxide ligands remaining from the starting material. Thus it would appear that the greater flexibility of the linear (L2)2− ligand [cf. cyclic analogue (L1)2−] allows for a chelating mode of coordination, even for Ti(IV), and that the simple synthesis/isolation of rac-L2Ti(OiPr)2 from wholly racemic precursors offers a convenient route to C2 symmetric geometrically cis complexes.
 |
| | Fig. 3 Structure of L2Ti(OiPr)2; hydrogen atoms have been omitted for clarity and ORTEP ellipsoids set at the 30% probability level. Important bond lengths (Å) and angles (°): Ti(1)–O(1) 1.906(2), Ti(1)–O(2) 1.901(2), Ti(1)–O(3) 1.836(2), Ti(1)–O(4) 1.834(2), Ti(1)–N(1) 2.325(2), Ti(1)–N(2) 2.313(2); O(1)–Ti(1)–O(2) 163.03(6), O(3)–Ti(1)–O(4) 107.70(7), N(1)–Ti(1)–N(2) 76.20(6). | |
The molecular structure of rac-L2Ti(OiPr)2 contains general structural features characteristic of Ti(IV) complexes of chelating bis(tertiary amine) bis(phenolate) ligands,4 and closely resembles the complexes LCF3MX2 (M = Ti, X = Cl; M = Zr, X = CH2Ph) recently synthesized by Carpentier and co-workers containing a diaminodialkoxo ligand system derived from the related but achiral diol [HOC(CF3)2CH2N(Me)CH2]2 (H2LCF3).6b Thus the transoid O(1)–Ti(1)–O(2) and cisoid N(1)–Ti(1)–N(2) angles associated with the L2Ti unit [163.03(6) and 76.20(6)°, respectively] and the related Ti–O and Ti–N distances [1.903 (mean) and 2.319 Å (mean)] are essentially identical to the corresponding parameters for LCF3TiCl2.6b Encouraged by the reported synthesis of complexes of the type LCF3ZrX2 we also examined the reactivity of H2L2 towards Zr(IV) precursors. Reaction with zirconium tetrakis(propoxide), however, yields the 1 : 2 complex, Zr(L2)2 irrespective of reaction stoichiometry, which crystallizes from hexane solution with both L2 ligands of meso (R,S) stereochemistry (Fig. 4). Similar reactivity to generate eight coordinate Zr(IV) complexes of the type ZrL2 is well precedented,13 with slight lengthening in the Zr–O [2.056(2)–2.073(1) Å] and Zr–N bonds [2.595(2)–2.621(2) Å] with respect to LCF3Zr(CH2Ph)2 presumably reflecting increases in steric crowding at the metal centre.6b
 |
| | Fig. 4 (a) Structure of L22Zr; hydrogen atoms have been omitted for clarity and ORTEP ellipsoids set at the 30% probability level. Important bond lengths (Å) and angles (°): Zr(1)–O(1) 2.060(2), Zr(1)–O(2) 2.057(2), Zr(1)–O(3) 2.073(2), Zr(1)–O(4) 2.056(2), Zr(1)–N(1) 2.620(2), Zr(1)–N(2) 2.621(2), Zr(1)–N(3) 2.595(2), Zr(1)–N(4) 2.616(2); O(1)–Ti(1)–O(2) 88.99(6), O(3)–Ti(1)–O(4) 87.69(6), N(1)–Ti(1)–N(2) 67.43(6), N(3)–Ti(1)–N(4) 67.62(6); (b) structure of L22Zr emphasizing the coordination geometry at the metal centre. | |
Conclusions
Epoxide ring opening chemistry using 1,4-diazacycloheptane (or its cyclic six-membered analogue 1,4-diazacyclohexane), or the related linear species N,N′-dimethylethylenediamine, and racemic (±)-3,3-dimethyl-1,2-epoxybutane gives single-step access to pendant alcohol functionalised ditertiary amine pro-ligands. Thus, [HOC(H)tBuCH2N(R)CH2]2 [R2 = CH2CH2CH2 (H2L1) and CH2CH2] can be isolated as diastereomerically pure crystalline materials from the reaction mixture, while oily H2L2 (R2 = Me2) is obtained as a mixture of rac and meso isomers. The contrasting reactions of H2L1 and H2L2 towards homoleptic group 4 alkoxides highlight the crucial role of ligand backbone flexibility in complex formation. Thus, the chemistry of the more conformationally rigid system (L1)2− appears to be constrained by the cyclic ligand core, such that it adopts a bridging (μ2:η2,η2) mode of coordination towards Ti(IV), leading to dinuclear metal systems [e.g.L1Ti2(OiPr)6]. By contrast, the more flexible linear system (L2)2− binds to both Ti(IV) and Zr(IV) in a chelating fashion leading, for example, to the syntheses of the mononuclear 1 : 1 complex L2Ti(OiPr)2 and the 1 : 2 Zr(IV) complex Zr(L2)2. Although H2L2 is necessarily employed as a mixture of rac and meso diastereomers in its reaction with Ti(OiPr)4, simple crystallization yields solely the C2 symmetric isomer of L2Ti(OiPr)2, featuring the rac form of the ligand. A simple procedure for the synthesis and isolation of diastereomerically pure C2 symmetric Ti(IV) complexes of cis geometry from racemic precursors is therefore presented.
Acknowledgements
We acknowledge the EPSRC and Johnson Matthey for funding this project, and the assistance of the EPSRC National Mass Spectrometry Service Centre (Swansea).
References
- For recent reviews, see:
(a) P. Corradini, G. Guerra and L. Cavallo, Acc. Chem. Res., 2004, 37, 231 CrossRef CAS;
(b) J. Gromada, A. Mortreux and J.-F. Carpentier, Coord. Chem. Rev., 2004, 248, 397 CrossRef;
(c) Y. Suzuki, H. Terao and T. Fujita, Bull. Chem. Soc. Jpn., 2003, 76, 1493 CrossRef CAS;
(d) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283 CrossRef CAS;
(e) G. W. Coates, J. Chem. Soc., Dalton Trans., 2002, 467 RSC;
(f) G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428 CrossRef CAS.
-
(a) L. G. Hubert-Pfalzgraf, Coord. Chem. Rev., 1998, 178–180, 967 CrossRef CAS;
(b) R. M. Mehrotra and A. Singh, Prog. Inorg. Chem., 1997, 46, 239;
(c)
D. C. Bradley, R. M. Mehrotra, I. P. Rothwell and A. Singh, in Alkoxo and Aryloxo Derivatives of Metals, Academic Press, London, 2001 Search PubMed.
- For examples of sterically encumbered chelating bis(aryloxide) ligands, see:
(a) S. Fokken, T. P. Spaniol, J. Okuda, F. G. Sernetz and R. Mullhaupt, Organometallics, 1997, 16, 4240 CrossRef CAS;
(b) A. van der Linden, C. J. Schaverien, N. Meijboom, C. Ganter and A. G. Orpen, J. Am. Chem. Soc., 1995, 117, 3008 CrossRef CAS.
- For recent examples of chelating bis(aryloxide) ligands featuring additional donor centres, see:
(a) A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Chem. Commun., 2006, 887 Search PubMed;
(b) C. Capacchione, R. Manivannan, M. Barone, K. Beckerle, R. Centore, L. Oliva, A. Proto, A. Tuzi, T. P. Spaniol and J. Okuda, Organometallics, 2005, 24, 2971 CrossRef CAS;
(c) C. L. Boyd, T. Toupance, B. R. Tyrrell, B. D. Ward, C. R. Wilson, A. R. Cowley and P. Mountford, Organometallics, 2005, 24, 309 CrossRef CAS;
(d) S. Segal, I. Goldberg and M. Kol, Organometallics, 2005, 24, 200 CrossRef CAS;
(e) L. S. Natrajan, C. Wilson, J. Okuda and P. L. Arnold, Eur. J. Inorg. Chem., 2004, 3724 CAS;
(f) P. D. Knight, I. J. Munslow, P. N. O’Shaughnessy and P. Scott, Chem. Commun., 2004, 894 RSC;
(g) C. Capacchione, F. De Carlo, C. Zannoni, J. Okuda and A. Proto, Macromolecules, 2004, 37, 8918 CrossRef CAS;
(h) S. Groysman, I. Goldberg, M. Kol, E. Genizi and Z. Goldschmidt, Organometallics, 2003, 22, 3013 CrossRef CAS;
(i) S. Groysman, I. Goldberg, M. Kol, E. Genizi and Z. Goldschmidt, Inorg. Chim. Acta, 2003, 345, 137 CrossRef CAS;
(j) V. Reimer, T. P. Spaniol, J. Okuda, H. Ebeling, A. Tuchbreiter and R. Mülhaupt, Inorg. Chim. Acta, 2003, 345, 221 CrossRef CAS;
(k) C. Capacchione, A. Proto, H. Ebeling, R. Mülhaupt, K. Möller, T. P. Spaniol and J. Okuda, J. Am. Chem. Soc., 2003, 125, 4964 CrossRef CAS;
(l) Y. Nakayama, H. Bando, Y. Sonobe, H. Kaneko, N. Kashiwa and T. Fujita, J. Catal., 2003, 215, 171 CrossRef CAS;
(m) E. Y. Tshuva, S. Groysman, I. Goldberg, M. Kol and Z. Goldschmidt, Organometallics, 2002, 21, 662 CrossRef CAS;
(n) S. Fokken, F. Reichwald, T. P. Spaniol and J. Okuda, J. Organomet. Chem., 2002, 663, 158 CrossRef CAS;
(o) M. C. W. Chan, K.-H. Tam, Y.-L. Pui and N. Zhu, J. Chem. Soc., Dalton Trans., 2002, 3085 RSC;
(p) S. Matsui and T. Fujita, Catal. Today, 2001, 66, 61 CrossRef;
(q) E. Y. Tshuva, I. Goldberg, M. Kol and Z. Goldschmidt, Organometallics, 2001, 20, 3017 CrossRef CAS;
(r) S. Y. Bylikin, D. A. Robson, N. A. H. Male, L. H. Rees, P. Mountford and M. Schröder, J. Chem. Soc., Dalton Trans., 2001, 170 RSC;
(s) E. Y. Tshuva, I. Goldberg, M. Kol and Z. Goldschmidt, Inorg. Chem. Commun., 2000, 3, 611 CrossRef CAS;
(t) E. Y. Tshuva, I. Goldberg, M. Kol, H. Weitman and Z. Goldschmidt, Chem. Commun., 2000, 379 RSC;
(u) E. Y. Tshuva, I. Goldberg and M. Kol, J. Am. Chem. Soc., 2000, 122, 10706 CrossRef CAS.
- For recent examples of aryloxide ligands featuring Schiff base or related pendant donors, see:
(a) I. Westmoreland, I. J. Munslow, A. J. Clarke, G. Clarkson, R. J. Deeth and P. Scott, J. Organomet. Chem., 2006, 691, 2228 CrossRef CAS;
(b) R. K. J. Bott, M. Mammond, P. N. Horton, S. J. Lancaster, M. Bochmann and P. Scott, Dalton Trans., 2005, 3611 RSC;
(c) D. A. Pennington, S. J. Coles, M. B. Hursthouse, M. Bochmann and S. J. Lancaster, Chem. Commun., 2005, 3150 RSC;
(d) M. Sanz, T. Cuenca, M. Galakhov, A. Grassi, R. K. J. Bott, D. L. Hughes, S. J. Lancaster and M. Bochmann, Organometallics, 2004, 23, 5324 CrossRef CAS;
(e) C. Cuomo, M. Strianese, T. Cuenca, M. Sanz and A. Grassi, Macromolecules, 2004, 37, 7469 CrossRef CAS;
(f) A. F. Mason and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 16326 CrossRef CAS;
(g) D. C. H. Oakes, B. S. Kimberley, V. C. Gibson, D. J. Jones, A. J. P. White and D. J. Williams, Chem. Commun., 2004, 2174 RSC;
(h) P. D. Knight, P. N. O’Shaughnessy, I. J. Munslow, B. S. Kimberley and P. Scott, J. Organomet. Chem., 2003, 683, 103 CrossRef CAS;
(i) D. Owiny, S. Parkin and F. T. Lapido, J. Organomet. Chem., 2003, 678, 134 CrossRef CAS;
(j) S. Reinartz, A. F. Mason, E. B. Lobkovsky and G. W. Coates, Organometallics, 2003, 22, 2542 CrossRef CAS;
(k) R. Furuyama, J. Saito, S. Ishii, M. Mitani, S. Matsui, Y. Tohi, H. Makio, N. Matsukawa, H. Tanaka and T. Fujita, J. Mol. Catal. A: Chem., 2003, 200, 31 CrossRef CAS;
(l) P. D. Hustad and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 11578 CrossRef CAS;
(m) P. D. Knight, A. J. Clarke, B. S. Kimberley, R. A. Jackson and P. Scott, Chem. Commun., 2002, 352 RSC;
(n) S. Matsui, M. Mitani, J. Saitoo, Y. Tohi, H. Makio, N. Matsukawa, Y. Takagi, K. Tsuru, M. Nitabaru, T. Nakano, H. Tanaka, N. Kasiwa and T. Fujita, J. Am. Chem. Soc., 2001, 123, 684;
(o) M. Wang, H. Zhu, K. Jin, D. Dai and L. Sun, J. Catal., 2001, 220, 392;
(p) J. Balsells, P. J. Carroll and P. J. Walsh, Inorg. Chem., 2001, 40, 5568 CrossRef CAS;
(q) P. R. Woodman, N. W. Alcock, I. J. Munslow, C. J. Sanders and P. Scott, J. Chem. Soc., Dalton Trans., 2000, 3340 RSC.
- For recent examples of alkoxide ligands featuring pendant donors, see:
(a) M. E. G. Skinner, T. Toupance, D. A. Cowhig, B. R. Tyrrell and P. Mountford, Organometallics, 2005, 24, 5586 CrossRef CAS;
(b) L. Lavanant, T.-Y. Chou, Y. Chi, C. W. Lehmann, L. Toupet and J.-F. Carpentier, Organometallics, 2004, 23, 5450 CrossRef CAS;
(c) I. J. Munslow, A. J. Clarke, R. J. Deeth, I. Westmoreland and P. Scott, Chem. Commun., 2002, 1868 RSC;
(d) R. Manivannan and G. Sundararajan, Macromolecules, 2002, 35, 7883 CrossRef CAS;
(e) Y. Kim, Y. Han and Y. Do, J. Organomet. Chem., 2001, 634, 19 CrossRef CAS;
(f) D. A. Robson, S. Y. Bylikin, M. Cantuel, N. A. H. Male, L. H. Rees, P. Mountford and M. Schröder, J. Chem. Soc., Dalton Trans., 2001, 157 RSC;
(g) D. A. Robson, L. H. Rees, P. Mountford and M. Schröder, Chem. Commun., 2000, 1269 RSC;
(h) R. Manivannan, G. Sundararajan and W. Kaminsky, J. Mol. Catal. A: Chem., 2000, 160, 85 CrossRef CAS;
(i) R. M. Gauvin, J. A. Osborn and J. Kress, Organometallics, 2000, 19, 2944 CrossRef CAS;
(j) P. Shao, R. A. L. Gendron, D. J. Berg and G. W. Bushnell, Organometallics, 2000, 18, 509 CrossRef.
- P. D. Knight and P. Scott, Coord. Chem. Rev., 2003, 242, 125 CrossRef CAS.
-
R. R. Strevens and I. A. Fallis, manuscript in preparation.
-
(a) DENZO in: Z. Otwinowski and W. Minor, Methods Enzymol., 1996, 276, 307 Search PubMed;
(b) Collect: Data Collection Software, R. Hooft, Nonius B. V., 1998;
(c) G. M. Sheldrick, Acta Crystallogr., Sect. A, 1990, 46, 467 CrossRef;
(d)
G. M. Sheldrick, University of Göttingen, 1997;
(e) Sortav: R. H. Blessing, Acta Crystallogr., Sect. A, 1995, 51, 33 Search PubMed.
-
R. E. Baghurst, PhD thesis, Cardiff University, 2005.
-
(a) S. D. Bull, M. G. Davidson, A. L. Johnson, D. E. J. E. Robinson and M. F. Mahon, Chem. Commun., 2003, 1750 RSC;
(b) M. Kol, M. Shamis, I. Goldberg, Z. Goldschmidt, S. Alfi and E. Hayut-Salant, Inorg. Chem. Commun., 2002, 4, 177;
(c) Y. Kim and J. G. Verkade, Organometallics, 2002, 21, 2395 CrossRef.
-
J. Emsley in, The Elements, Oxford University Press, Oxford, 1995 Search PubMed.
- For a recent example featuring a bis(N-donor) bis(aryloxide) ligand system, see: T. Toupance, S. R. Dubberley, N. H. Rees, B. R. Tyrrell and P. Mountford, Organometallics, 2002, 21, 1367 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and characterizing data for N,N′-bis(2-hydroxy-3,3-dimethylbutyl)-1,4-diazacyclohexane; full details of the crystal structures of L1Ti2(OiPr)6, L12Ti2(OEt)2(μ-O), L2Ti(OiPr)2 and L22Zr; and 13C NMR spectra relevant to the separation of rac- and meso-L2Ti(OiPr)2. See DOI: 10.1039/b608680b |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2007 |
Click here to see how this site uses Cookies. View our privacy policy here.