Substitution of allylic acetates with sodium para-toluenesulfinate in aqueous media using allylpalladium chloride dimer and a water-soluble ligand as the catalytic system; electrospray ionisation mass spectrometry analysis†
Carole
Chevrin
a,
Jean
Le Bras
a,
Anna
Roglans
b,
Dominique
Harakat
a and
Jacques
Muzart
*a
aUnité Mixte de Recherche 6519 “Réactions Sélectives et Applications”, Boîte no 44, CNRS-Université de Reims Champagne-Ardenne, BP 1039, 51687 Reims cedex 2, France. E-mail: jacques.muzart@univ-reims.fr; Fax: +33 3-2691-3166; Tel: +33 3-2691-3237
bDepartment of Chemistry, Universitat de Girona, Campus de Montilivi, 17071 Girona, Spain
First published on 16th November 2006
Abstract
The allylic substitution of allylic acetates by sodium para-toluenesulfinate in aqueous media was catalyzed by [(η3-allyl)PdCl]2 associated with [(HOCH2CH2NHCOCH2)2NCH2]2. High yields could be obtained but the recycling of the catalytic system proved to be weakly effective. ESI-MS analysis has led to the suggestion of a possible catalytic cycle involving a PdIV intermediate.
1. Introduction
Nowadays, chemistry that takes place in water receives considerable attention for environmental, economic and safety reasons, and also for showing unique reactivities and selectivities that are not seen in organic solvents.1 In recent years, we have been involved in this research topic.2–5 In particular, we have disclosed the water-promoted allylic substitution of (E)-1-acetoxy-1,3-diphenylpropene (1) by various nucleophiles at 50 °C in the absence of palladium complexes3 (eqn. (1)) and, with acetylacetonate anion as the nucleophilic species, the unusual role of PdCl2(MeCN)2/LH in the formation of PhCH(CH(COMe)2)CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh4 (eqn. (2) (LH = [(HOCH2CH2NHCOCH2)2NCH2]2). The allylic substitution of (E)-3-phenyl-1-para-tolylallyl acetate by C-, O-, S- and N-nucleophiles also occurred effectively in the absence of a metal catalyst3 but the reactivity of (E)-4-phenylbut-3-en-2-yl acetate (2) was much lower.6 The results obtained from 2 and a range of other allylic acetates are described here, with an electrospray ionisation mass spectrometry (ESI-MS) investigation suggesting mechanistic possibilities.
CHPh4 (eqn. (2) (LH = [(HOCH2CH2NHCOCH2)2NCH2]2). The allylic substitution of (E)-3-phenyl-1-para-tolylallyl acetate by C-, O-, S- and N-nucleophiles also occurred effectively in the absence of a metal catalyst3 but the reactivity of (E)-4-phenylbut-3-en-2-yl acetate (2) was much lower.6 The results obtained from 2 and a range of other allylic acetates are described here, with an electrospray ionisation mass spectrometry (ESI-MS) investigation suggesting mechanistic possibilities. | (1) |
 | (2) |
2. Results and discussion
Treating 2 with 1.1 equiv. of sodium para-toluenesulfinate (3) in a 1 : 1 mixture of H2O and DMF at 100 °C for 24 h led to a 55% conversion of 2, with the appearance of 4-phenylbut-3-en-2-ol (4) (33%), and adducts 5 and 6 (20%, 5/6 ratio = 1 : 1) (eqn. (3)). With 2 equiv. of 3, complete conversion of 2 took place and the yield of the adducts increased to 41%. Decreasing the reaction temperature afforded lower conversions, as exemplified in Fig. 1; no reaction occurring below 60 °C. At 100 °C, switching from 3 to morpholine or CH2(COMe)2/K2CO3 as the nucleophilic species led only to the formation of 4. | (3) |
 | ||
| Fig. 1 Influence of reaction temperature on the conversion of 2 in the absence of a Pd catalyst. Experimental conditions: 2 (0.8 mmol), 3 (2 equiv.), H2O (1 mL), DMF (1 mL), 3 h. | ||
The above low selectivities and reactivities led us to examine the influence of a Pd catalyst using our previously reported aqueous conditions.3,4 In contrast to the results produced with 1 as the substrate, the nucleophilic addition of 3 to 2 did not occur at 50 °C using PdCl2(MeCN)2/LH as the catalyst and H2O/MeOH as the solvent. Gratifyingly though, a mixture of [(η3-allyl)PdCl]2 (7) and LH proved to be an efficient and selective catalytic system in H2O/MeOH, even at 50 °C, affording a 98% yield of 5 in 3 h (Table 1, entry 1). Similar yields were obtained using DMF, MeCN and HMPA as the co-solvent, while DMSO and THF were less effective (Fig. 2). A high (98%) selectivity was also observed using only water as solvent but the conversion dropped to 68%. As above, the substitution of 2 by morpholine, or the sodium salt of acetylacetone or dimethylmalonate did not occur under these Pd-catalyzed conditions.
| Entry | Substrate | Time/h | Product | Yield (%) |
|---|---|---|---|---|
| 1 |

|
3 |

|
98 |
| 2 |

|
3 |

|
99 |
| 3 |

|
3 |

|
99 |
| 4 |

|
24 |
|
99 |
| 5 |

|
48 |

|
22 |
| 6 |

|
24 |
|
59 |
| 7 |

|
22 |

|
55 |
![Reaction of 2 with 3 using [(η3-allyl)PdCl]2/LH as the catalyst in various aqueous mixtures. Experimental conditions: 2 (1 mmol), 3 (2 equiv.), 7 (0.01 equiv.), LH (0.02 equiv.), H2O (1 mL), co-solvent (1 mL), 50 °C, 3 h.](/image/article/2007/NJ/b613562e/b613562e-f2.gif) | ||
| Fig. 2 Reaction of 2 with 3 using [(η3-allyl)PdCl]2/LH as the catalyst in various aqueous mixtures. Experimental conditions: 2 (1 mmol), 3 (2 equiv.), 7 (0.01 equiv.), LH (0.02 equiv.), H2O (1 mL), co-solvent (1 mL), 50 °C, 3 h. | ||
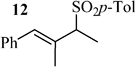
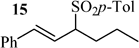
Following these observations, a range of allylic acetates were subjected to a 7/LH-catalyzed reaction with 3 in H2O/MeOH. As shown by the results collected in Table 1, the substitution occurred in all cases on the less crowded carbon. The fact that isomeric allylic acetates 11 and 13 afford the same product (12; Table 1, entries 4 and 5) indicates that 11 and 13 operate through the same allylic intermediate. Substitution of the central atom of the allylic moiety strongly decreased the reaction rate (Table 1, entries 1 and 4). According to the high difference of reactivity between 11 and 13 (Table 1, entries 4 and 5), it also appears that the facility to form the reactive intermediate greatly depends on the structure of the substrate.
A point of interest in metal-catalyzed reactions in aqueous media is the possibility of recycling. To clarify this issue, a reaction carried out as indicated in Table 1, entry 1, was extracted with CH2Cl2. After addition of MeOH, 2 and 3 to the aqueous phase, followed by heating at 50 °C, 5 h was required for the complete consumption of 2, providing 5 in 98% yield. A second recycling was much more sluggish, affording no more than 40% conversion in 24 h.
To obtain information about the mechanism of these reactions, an ESI-MS analysis was undertaken.7 Each component of the catalytic system was first analyzed. Because Pd and Cl display 6 and 2 isotopes, respectively, the ions containing these isotopes should be mass-detected as clusters of isotopomeric ions whose center depends on the most abundant isotope (106 for Pd and 35 for Cl).
The ESI(+)-MS of 7 in a MeOH/H2O/THF (5 : 5 : 1) mixture showed three main peaks at m/z = 389, 461 and 513, corresponding to [(PdC3H5Cl)2 + Na]+, [(PdC3H5Cl)2 + THF + Na]+ and the trinuclear metal cluster [(PdC3H5Cl)3 – Cl]+, respectively (Fig. S1, ESI†). Two cationic forms of LH at m/z = 465 ([LH + H]+) and 487 ([LH + Na]+) were observed from the ESI(+)-MS of LH in MeOH/H2O (Fig. S2, ESI†). The ESI(+)-MS of an 1 : 2 mixture of 7 and LH in MeOH/H2O revealed a peak at m/z = 611 corresponding to [LHPdC3H5]+ (Fig. S3, ESI†). The addition of allylic acetate 2 (2.0 equiv. with respect to 7) to this 7 + LH mixture did not afford new peaks. The ESI(+)-MS spectra only evolved after the addition of sulfinate 3 (4 equiv./7); after one hour at 50 °C, besides the previous [LHPdC3H5]+ peak, peaks at m/z = 701 and 879 were obtained. Peak m/z = 701 is attributable to [LHPd(2) – OAc]+ while m/z = 879 is consistent with [LHPd(2)SO2p-Tol – OAc + Na]+ and [LHPd(5) + Na]+ (Fig. 3 and Fig. 4). After 24 h, the mixture was extracted with CH2Cl2 and the ESI(+)-MS spectrum of the aqueous phase recorded. This spectrum showed the peak at m/z = 611 already observed, and two new clusters centered at m/z = 747 and 769, attributable to [LHPdSO2p-Tol – H + Na]+ and [LHPdSO2p-Tol – 2H + 2Na]+ (Fig. 5; Fig. S4, ESI†).
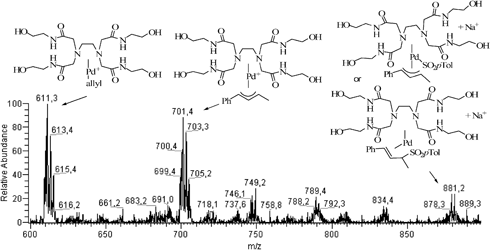 | ||
| Fig. 3 ESI(+)-MS spectrum of the crude mixture obtained under the following conditions: 7, LH (2 equiv.), 2 (2 equiv.), 3 (4 equiv.), H2O/MeOH (1 : 1), 50 °C, 1 h. | ||
 | ||
| Fig. 4 Expanded ESI(+)-MS spectra of detected species from Fig. 3 and corresponding theoretical spectra. | ||
 | ||
| Fig. 5 ESI(+)-MS spectrum of the aqueous phase after extraction of the crude mixture with CH2Cl2. Experimental conditions: As in Fig. 3 for 24 h followed by extraction with CH2Cl2. | ||
Further ESI-MS experiments were carried out using either CH2(COMe)2 + K2CO3 or NaCH(COMe)2. In the first case (Fig. S5, ESI†), we only observed [LHPdC3H5]+ and [LHPdC3H5 – H + K]+ clusters. The use of NaCH(COMe)2 led similarly to [LHPdC3H5]+ and [LHPdC3H5– H + Na]+ clusters but, in addition, to traces of [LHPd(2) – OAc]+ (Fig. S6, ESI†). These ESI-MS analyses, which are in agreement with the reactivity of the sulfinate anion and the reluctant addition of acetylacetonate anion highlighted in the preparative experiments, have lead us to make mechanistic proposals for the allylic substitution of 2 by 3 under these particular Pd-catalyzed conditions.
Mixing 7 with LH led to a monomeric η3- or η1-allylpalladium complex, having LH as a bidentate ligand, namely LHPd(C3H5)Cl, revealed by the cluster [LHPdC3H5]+. This complex is either cationic with an η3-allyl moiety, or neutral with an η1-allyl moiety (Scheme 1). Let us now to consider two possible mechanisms, A and B, that could explain the reactivity of these species in the presence of 3 and 7.
 | ||
| Scheme 1 | ||
Mechanism A
Mechanism A is similar to the catalytic cycle usually described for Tsuji–Trost reactions using a mixture of 7 and a phosphine as the catalyst (Scheme 2). Pd0 complex A1 arises from the nucleophilic addition of 3 to LHPd(C3H5)Cl. An exchange of ligand with 2 leads to A2 and then to cationic PdII complex A3. The reaction of 3 with A3 affords Pd0 complex A4, which evolves to A2, closing the catalytic cycle. | ||
| Scheme 2 | ||
Intermediates A2 and A3 are consistent with the cluster at m/z = 701, attributed to [LHPd(2) – OAc]+, while A4 is in agreement with the cluster at m/z = 879. In contrast, mechanism A is not in accordance with the ESI-MS spectrum recorded at the end of the reaction, which highlights a cluster at m/z = 611 corresponding to the complex LHPd(C3H5)X (X = Cl, OAc or SO2p-Tol). To accept mechanism A, it is a required consideration that only a slight portion of LHPd(C3H5)Cl reacts with 3. LHPd(C3H5)Cl would act as a reservoir of PdII, which is reduced to Pd0 to enter the catalytic cycle. However the need for an explanation remains as to why 3 would react with A3 rather than with LHPd(C3H5)Cl.
Since 2 equiv. per Pd of polydentate ligand LH are used, according to the research groups of Shaw and Jutand,8–11 the most likely form of the allyl moiety of a species such as LHPd(C3H5)Cl is the η1-allyl one.12 Consequently, this species would be the neutral complex LHPdCl(η1-allyl) (Scheme 1). As a cationic palladium complex is more reactive towards nucleophilic addition than a neutral one,10 this could explain the reaction of 3 with the cationic complex A3 rather than with LHPdCl(η1-allyl).
Mechanism B
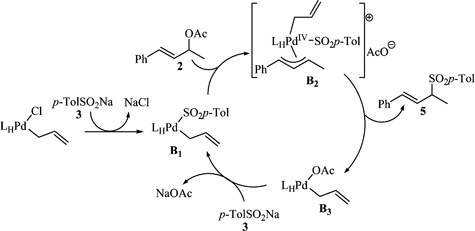
For mechanism B, we take into consideration (i) the most likely structure of LHPd(C3H5)Cl,8–11i.e. the neutral complex LHPdCl(η1-allyl), (ii) the reported substitution at Pd of chloropalladium complexes with PhSO2Na,13 and (iii) Kurosawa’s proposal of a reductive elimination leading to an R-allyl from PdIV(allyl)2R(X)PPh3.14An exchange of anion between LHPdCl(η1-allyl) and 3 affords B1 (Scheme 3). Such a step could be considered a transmetallation. The reaction of B1 with 2 leads to cationic PdIV complex B2.15 Kurosawa et al. have shown that reductive elimination was more efficient from an η3-allyl than from an η1-allylpalladium complex.17–19 Consequently, reductive elimination from B2 produces 5 rather than allylSO2p-Tol. Such an elimination results in B3, which interacts with 3 to close the catalytic cycle.
 | ||
| Scheme 3 | ||
According to mechanism B, the real catalyst is LHPd(SO2p-Tol)(η1-allyl) (B1). B1 is consistent with the cluster at m/z = 611, corresponding to [LHPdC3H5]+, which has been observed in both the crude mixture and the aqueous extraction phase. PdIV complexes have been isolated20 but the formation of a PdIV complex from a PdII complex and an allylic acetate, i.e. in Scheme 3 the formation of (η1-allyl)(η3-allyl)PdIV species B2 from 2 and (η1-allyl)PdII intermediate B1, is an unusual step. Nevertheless, Kurosawa’s team have (i) already suspected a PdIV intermediate from the oxidative addition of allyl acetate to an (η3-allyl)PdII complex14 and (ii) shown that (η1-organo)metalII complexes are more similar to metal0 than metalII complexes.21 In addition, the formation of a cationic (η3-allyl)PdIV intermediate has recently been proposed for the reaction of a vinylpalladium chloride with an allylic alcohol in an aqueous medium.22
The clusters at m/z = 701, 747, 769 and 879 are not directly consistent with the intermediates of mechanism B but can be connected to B2 through the reactions depicted in Scheme 4. Indeed, the cluster at m/z = 701 corresponds to [LHPd(PhC3H3Me)]+ while those centered at m/z = 747 and 769 occur from [LHPdSO2p-Tol]+. These species would arise from reductive eliminations (paths (a)23 and (b)24) less favored than the one depicted in Scheme 3. The cluster at m/z = 879 can be produced not only from LHPd(5) (intermediate A4 in Scheme 2) but also from B2via the nucleophilic addition of acetate anion to the η1-allyl ligand (path (c)), i.e. a reaction already documented for a cationic (η1-allyl)palladium complex.25–27 Steps (a), (b) and (c) would produce minute amounts of organic compounds since the C3H5 moiety comes from the catalyst. Furthermore, step (c), which leads to allyl acetate, would be reversible.
 | ||
| Scheme 4 | ||
A possible explanation for the unreactivity of morpholine, and the sodium salts of acetylacetone and dimethylmalonate, involves the influence of steric effects.26,33 LH is a crowded ligand and, according to mechanism B, the sulfinate group is a ligand of palladium before being linked to the allyl moiety. Consequently, the size of the nucleophilic species could play a decisive role. Mechanisms A and B remain, nevertheless, hypothetical but we have, however, to point out that the extracted aqueous phase contains LHPdX(allyl). Such a species, namely B1, is directly involved in mechanism B, and we have obtained 5 in 98% yield from a preparative experiment using a recycled aqueous phase.
3. Conclusion
A mixture of both [(η3-allyl)PdCl]2 and a water soluble ligand catalyzes the allylic substitution of allylic acetates by sodium para-toluenesulfinate in a variety of aqueous media, but the system quickly looses its catalytic properties upon recycling. Substitution of the allylic moiety of the substrate has a significant influence on the efficiency of the reaction. ESI-MS analyses have led to the suggestion of a possible catalytic cycle involving a PdIV intermediate.4. Experimental
3.1. General procedure for Pd-catalyzed substitution
To a mixture of [(η3-allyl)PdCl]2 (3.6 mg, 0.01 mmol) and [(HOCH2CH2NHCOCH2)2NCH2]2 (9.3 mg, 0.02 mmol) in MeOH (1 mL), stirred at room temperature for 1 h, was added the substrate (1.0 mmol), sodium para-toluenesulfinate (388 mg, 2 mmol) and H2O (1 mL). After heating at 50 °C for the time indicated in Table 1, the organic compounds were extracted with CH2Cl2. The organic layer was dried over MgSO4 and concentrated to give the crude product. This product was purified by chromatography over silica gel eluted with a 10 : 90 mixture of AcOEt and petroleum ether.3.2. ESI-MS analysis
ESI-MS analyses were recorded on a Navigator quadrupole mass spectrometer (Finnigan AQA ThermoQuest) equipped with an electrospray ion source. The instrument was operated either in the positive ESI(+) or negative ESI(–) ion mode at a probe tip voltage of 3 kV. Samples were introduced into the mass spectrometer ion source directly through a Rheodyne injector with a 20 µL sample loop. The mobile phase flow (100 µL min–1 of 70 : 30 v/v MeOH/H2O) was delivered by a P2000 HPLC pump (ThermoQuest) to the vaporization nozzle of the electrospray ion source (165 °C for ESI(+) and 140 °C for ESI(–)) and nitrogen was employed as both a drying and nebulizing gas. Skimmer cone voltages were varied between 10 and 100 eV. Spectra were typically an average of 15–20 scans. Theoretical isotope patterns were calculated using the Isoform program and were used to aid assignment. Association of the substrate with ions was common. [M + H]+ and [M + Na]+ were often observed, the latter appeared from reactions with traces of cations present, even in HPLC-grade solvent.7Acknowledgements
We are indebted to “Région Champagne-Ardenne” for a PhD studentship to C. C. Financial support from CNRS, PAI (Picasso), the MEC of Spain (project CTQ2005-04968) and the CIRIT-Generalitat de Catalunya (project 2005SGR00305) is gratefully acknowledged.References
- C.-J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68 RSC.
- (a) J. Le Bras and J. Muzart, Tetrahedron Lett., 2002, 43, 431 CrossRef CAS; (b) J. Le Bras and J. Muzart, Tetrahedron: Asymmetry, 2003, 14, 1911 CrossRef CAS.
- C. Chevrin, J. Le Bras, F. Hénin and J. Muzart, Tetrahedron Lett., 2003, 44, 8099 CrossRef CAS.
- C. Chevrin, J. Le Bras, F. Hénin, J. Muzart, A. Pla-Quintana, A. Roglans and R. Pleixats, Organometallics, 2004, 23, 4796 CrossRef CAS.
- (a) C. Chevrin, J. Le Bras, F. Hénin and J. Muzart, Synthesis, 2005, 2615 CAS; (b) M. Fousteris, C. Chevrin, J. Le Bras and J. Muzart, Green Chem., 2006, 8, 522 RSC.
- C. Chevrin, PhD Thesis, Université de Reims Champagne-Ardenne, Reims, France, 2005 Search PubMed.
- For a monograph on ESI-MS, see: Electrospray Ionization Mass Spectrometry, Fundamentals, Instrumentation, and Applications, ed. R. B. Cole, Wiley, New York, 1997 Search PubMed. For the application of ESI-MS to inorganic and organometallic chemistry, see the following reviews and references cited therein: R. Colton, A. D’Agostino and J. C. Traeger, Mass Spectrom. Rev., 1995, 14, 79 Search PubMed; W. Henderson, B. K. Nicholson and L. J. McCaffrey, Polyhedron, 1998, 17, 4291 Search PubMed; J. C. Traeger, Int. J. Mass Spectrom., 2000, 200, 387 Search PubMed; D. A. Plattner, Int. J. Mass Spectrom., 2001, 207, 125 CAS . For a recent review about the investigation of chemical reactions using this technique, see the following review and references cited therein: L. S. Santos, L. Knaack and J. O. Metzger, Int. J. Mass Spectrom., 2005, 246, 84 CrossRef CAS.
- J. Powell and B. L. Shaw, J. Chem. Soc. A, 1967, 1839 RSC.
- C. Amatore, A. Jutand, M. A. M’Barki, G. Meyer and L. Mottier, Eur. J. Inorg. Chem., 2001, 873 CrossRef CAS.
- T. Cantat, E. Génin, C. Giroud, G. Meyer and A. Jutand, J. Organomet. Chem., 2003, 687, 365 CrossRef CAS.
- A. Jutand, Eur. J. Inorg. Chem., 2003, 2017 CrossRef CAS.
- In contrast, the addition of 1 equiv. per Pd of a monodentate ligand to 7 led to the corresponding monomeric neutral η3-allyl complex: (a) J. Powell and B. L. Shaw, J. Chem. Soc. A, 1967, 1839 RSC; (b) K. Vrieze, A. P. Praat and P. Cossee, J. Organomet. Chem., 1968, 12, 533 CrossRef CAS; (c) P. Kisanga and R. A. Widenhoefer, J. Am. Chem. Soc., 2000, 122, 10017 CrossRef CAS; (d) L. I. Rodríguez, O. Rossell, M. Seco, A. Grabulosa, G. Muller and M. Rocamora, Organometallics, 2006, 25, 1368 CrossRef CAS.
- (a) B. Chiswell and L. M. Venanzi, J. Chem. Soc. A, 1966, 1246 RSC; (b) I.-P. Lorenz, E. Lindner and W. Reuther, Z. Anorg. Allg. Chem., 1975, 414, 30 CrossRef CAS; (c) M. N. Sokolov, R. Hernández-Molina, D. N. Dybtsev, E. V. Chubarova, S. F. Solodovnikov, N. V. Pervukhina, C. Vicent, R. Llusar and V. Fedin, Z. Anorg. Allg. Chem., 2002, 628, 2335 CrossRef CAS.
- H. Kurosawa, M. Emoto and A. Urabe, J. Chem. Soc., Chem. Commun., 1984, 968 RSC.
- A referee has pointed out that in a bis-allylpalladiumII complex, an unsubstituted allyl moiety more prefers η1-coordination than does a substituted example.16 Such a preference could also be the case for a bis-allylpalladiumIV complex.
- K. J. Szabó, Chem.–Eur. J., 2000, 6, 4413 CrossRef CAS.
- H. Kurosawa, M. Emoto, A. Urabe, K. Miki and N. Kasai, J. Am. Chem. Soc., 1985, 107, 8253 CrossRef CAS.
- H. Kurosawa, M. Emoto, H. Ohnishi, K. Miki, N. Kasai, K. Tatsumi and A. Nakamura, J. Am. Chem. Soc., 1987, 109, 6333 CrossRef CAS.
- H. Kurosawa, H. Ohnishi, M. Emoto, Y. Kawasaki and S. Murai, J. Am. Chem. Soc., 1988, 110, 6272 CrossRef CAS.
- (a) P. K. Byers, A. J. Canty, B. W. Skelton and A. H. White, J. Chem. Soc., Chem. Commun., 1986, 1722 RSC; (b) W. de Graaf, J. Boersma, W. J. J. Smeets, A. L. Spek and G. van Koten, Organometallics, 1989, 8, 2907 CrossRef CAS; (c) Y. Yamamoto, T. Ohno and K. Itoh, Angew. Chem., Int. Ed., 2002, 41, 3662 CrossRef CAS; (d) Y. Yamamoto, S. Kuwabara, S. Matsuo, T. Ohno, H. Nishiyama and K. Itoh, Organometallics, 2004, 23, 3898 CrossRef CAS; (e) J. Cámpora, P. Palma, D. del Río, J. A. López, E. Alvarez and N. G. Connelly, Organometallics, 2005, 24, 3624 CrossRef CAS; (f) A. R. Dick, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 12790 CrossRef CAS.
- H. Kurosawa, K. Ishii, Y. Kawasaki and S. Murai, Organometallics, 1989, 8, 1756 CrossRef CAS.
- J. Huang, L. Zhou and H. Jiang, Angew. Chem., Int. Ed., 2006, 45, 1945 CrossRef CAS.
- Allylsulfones have been obtained from the reductive elimination of the corresponding neutral (η3-allyl)(SO2p-Tol)palladium complexes: S. Kamijo, M. Al-Masum and Y. Yamamoto, Tetrahedron Lett., 1998, 39, 691 Search PubMed.
- The reductive elimination from a neutral (η1-allyl)(η3-allyl)palladium complex leading to 1,5-hexadiene has been reported: R. Bertani, A. Berton, G. Carturan and R. Campostrini, J. Organomet. Chem., 1988, 349, 263 Search PubMed.
- S. Bouquillon and J. Muzart, Eur. J. Org. Chem., 2001, 3301 CrossRef CAS.
- I. D. G. Watson and A. K. Yudin, J. Am. Chem. Soc., 2005, 127, 17516 CrossRef CAS.
- Nucleophilic addition onto (η1-allyl)PdCl(PPh3)2 is also documented,26,28 while the η1-moiety of neutral complexes such as (η1-allyl)PdArL229 and (η1-allyl)(η3-allyl)Pd(PPh3) reacts with electrophiles.16,30,31 A computational study on the reactivity of cationic (η1-allyl)Pd complexes has recently been reported32.
- B. Åkermark, G. Åkermark, L. S. Hegedus and K. Zetterberg, J. Am. Chem. Soc., 1981, 103, 3037 CrossRef.
- (a) H. Kurosawa and A. Urabe, Chem. Lett., 1985, 1839 CrossRef CAS; (b) N. Solin, J. Kjellgren and K. J. Szabó, Angew. Chem., Int. Ed., 2003, 42, 3656 CrossRef CAS.
- K. J. Szabó, Chem.–Eur. J., 2004, 10, 5269.
- C. Damez, B. Estrine, A. Bessmertnykh, S. Bouquillon, F. Hénin and J. Muzart, J. Mol. Catal. A: Chem., 2006, 244, 93 CrossRef CAS.
- M. García-Iglesias, E. Buñuel and D. J. Cárdenas, Organometallics, 2006, 25, 3611 CrossRef CAS.
- K. Tsurugi, N. Nomura and K. Aoi, Tetrahedron Lett., 2002, 43, 469 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Observed electrospray mass spectra and calculated isotopic distribution of the detected species (Fig. S1–S5). See DOI: 10.1039/b613562e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2007 |