Tripodal (N-alkylated) CMP(O) and malonamide ligands: synthesis, extraction of metal ions, and potentiometric studies
Dominik
Jańczewski
a,
David N.
Reinhoudt
a,
Willem
Verboom
*a,
Elżbieta
Malinowska
b,
Mariusz
Pietrzak
b,
Clément
Hill
c and
Cécile
Allignol
c
aLaboratory of Supramolecular Chemistry and Technology, Mesa+ Research Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE, Enschede, The Netherlands. E-mail: w.verboom@utwente.nl; Fax: +31 53489 4645; Tel: +31 53489 2977
bDepartment of Analytical Chemistry, Faculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664, Warsaw, Poland
cCommissariat à l’Energie Atomique, CEA-Valrho, BP 17171, 30207, Bagnols-sur-Cèze, France
First published on 14th November 2006
Abstract
Tripodal ligands build on the C-pivot (9b–e, 13b–d, and 17a–d) and trialkylbenzene platforms (10a,b, 11, 12, 14a,b, and 18a,b) bearing (N-alkylated) carbamoylmethylphosphine oxide (CMPO), carbamoylmethylphosphonate (CMP), and malonamide moieties were synthesized. Extraction studies with Am3+ and Am3+ show that in general there is a positive influence of the N-alkyl substituents in C-pivot CMP(O) ligands on the D (distribution) coefficients. The trialkylbenzene CMPO ligands 10a,b, 11, and 12 have considerably larger D coefficients than the corresponding C-pivot analogues 9a–e, although hardly having any selectivity, while N-alkylation gives rise to smaller D coefficients. Although less effective the extraction behavior of the C-pivot CMP analogues 13b–d shows more or less the same trend as the corresponding CMPO ligands 9b–e upon substitution of the carboxamide N-atom with different alkyl chains. The different malonamide ligands 17a–d and 18a,b are bad extractants, while N-alkylation makes them even worse. Potentiometric studies of CMP(O) and malonamide ligands in polymeric membranes on Pb2+, Cu2+, Ca2+, Mg2+, Na+, and K+ salts revealed that N-alkyl substituents increase the stability constants of ion–ionophore complexes compared to unsubstituted ligands. In polymeric membrane electrodes the ligands induce a selectivity pattern that differs significantly from the so-called Hofmeister series, giving the highest selectivity coefficients for UO22+ among all examined cations (Pb2+, Cu2+, Ca2+, Mg2+, Na+, K+).
Introduction
The interests in nuclear energy fluctuates together with the fossil fuel prices, making the topic very hot nowadays. A particularly important and not sufficiently solved problem is the removal of highly radiotoxic actinides from HLLW (high level liquid waste) formed during the reprocessing of the spent nuclear fuel and to separate the actinides from the more abundant lanthanides.1 After separation, the actinides can be transformed to less dangerous and easier to handle isotopes by transmutation. The most promising way is extraction of the actinides with highly selective ionophores, however, the design and synthesis of ligands fulfilling industrial requirements for this process, is still a challenge.The approach to connect different chelating groups to preorganized scaffolds to improve both the extraction efficiency and the selectivity is already explored in the field of actinide/lanthanide separation for quite some time.2 The main stream of investigations is directed toward ligands bearing four coordinating arms like carbamoylmethylphosphine oxide (CMPO)and carbamoylmethylphosphonate (CMP) substituted calixarenes3 and resorcinarenes,4 or calixarene functionalized picolinamides.5 Despite the evidence that actinides and lanthanides coordinate with bidentate ligands as CMP(O)6 and malonamides7 in a 1 ∶ 3 stoichiometry (metal3+ : ligand), only a few examples of tripodal ligands8 based on the triphenoxymethane platform were described by the groups of Scott8a,b and Böhmer.8c Recently, we reported the synthesis and complexation behavior of CMP(O) functionalized C-pivot tripodal ligands.9 In these tripodal ligands six donor oxygen atoms are brought together to fill the coordination sphere of the metal resulting in 1 ∶ 1 complexes.
Simple CMPO bidentate ligands6,10 with short branched N-substituents like isobutyl at the amide moiety, lead in general to an improved distribution and minimization of a third phase formation between the aqueous and the organic layer. However, for multicoordinate preorganized ligands mainly N-unsubstituted ligands have been investigated, except one N-methylated CMPO calix[4]arene3e and two CMP(O) N-butyl substituted resorcinarenes.4a
The 1,3,5-trialkylbenzene platform is attractive for the construction of different types of receptors, because ligating sites at the 2, 4, and 6 positions are preorganized at the same face of the molecule.11 However, to the best of our knowledge, it has not been used for the preparation of actinide/lanthanide ligands.
In this manuscript we describe the synthesis and complexation with Am3+ and Eu3+ of C-pivot and trialkylbenzene platform-based tripodal ligands containing (N-alkyl substituted) CMP(O)6,10 and malonamide7,12 ligating groups. Special attention is paid to the influence of the N-alkyl substituents on the extraction behavior. The ion–ionophore stability constants of complexes formed by these ligands with selected cations as well as the influence of the structure of these ligands on the response of cation-selective electrodes with polymeric membranes doped with these ligands are presented.
Results and discussion
Synthesis
Starting from the known C-pivot nitrile113 a series of tripodal secondary amines 3b–e was prepared using a slightly modified reductive alkylation method as described in the literature (Scheme 1).14 C-Pivot nitrile 1 was reacted with a large excess of the appropriate primary amine at 8 bar of hydrogen in the presence of 10% Pd–C15 at room temperature to give the tripodal amines 3b–e in excellent yields. | ||
| Scheme 1 | ||
In addition to correct molecular ion peaks in the HR-FAB mass spectra, their formation followed from their 1H NMR spectra with characteristic triplets at 2.56–2.69 ppm for the –CH2NHR methylene group. Primary tripodal amine3a was synthesized according to the literature procedure.8d
Reaction of tris(bromomethyl)benzene 411b with excess of 3-pentylamine afforded amine 5 in quantitative yield. For ligands with a longer spacer between the platform and the ligating site the tris(aminoethyl)benzenes 7a,b and 8 were prepared (Scheme 2). Catalytic hydrogenation of tris(cyanomethyl)benzenes 6a16a and 6b16b in the presence of Raney-Co gave the corresponding tripodal amines 7a,b in almost quantitative yields. The reductive alkylation of 6b with 3-pentylamine required 100 bar of hydrogen and 70 °C to give tripodal amine 8 in 90% yield. Besides the correct molecular ion signals displayed in the HR-FAB spectra, the 1H NMR spectra of amines 5 and 8 show characteristic multiplets for NCH around 2.45 ppm and that of amines 7a,b signals for PhCH2CH2NH2 around 2.8 ppm.
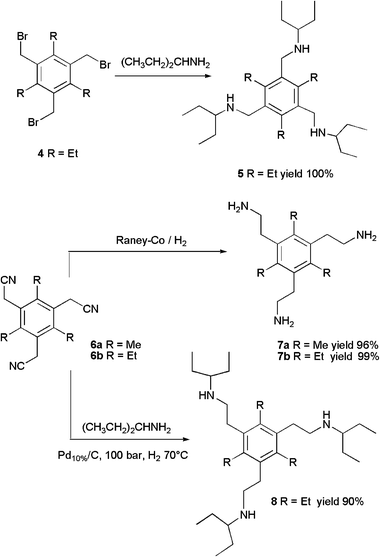 | ||
| Scheme 2 | ||
Carbamoylmethyl-phosphine oxide (CMPO) and -phosphonate (CMP) functions were introduced via reaction with chloroacetyl chloride in the presence of K2CO3, followed by an Arbuzov reaction with ethyl diphenyl phosphinite (for CMPO) or triethyl phosphite (for CMP), respectively, of the corresponding acetylated compounds (Schemes 3 and 4).17
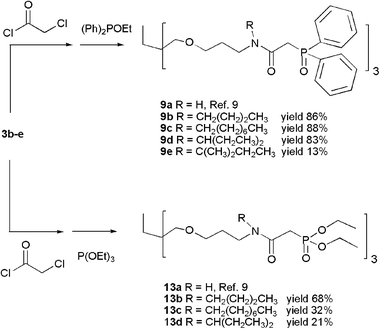 | ||
| Scheme 3 | ||
 | ||
| Scheme 4 | ||
The C-pivot based CMPO derivatives 9b–d were obtained in excellent yields. However in the case of the more crowded CMPO 9e the yield was only 13%, probably due to steric hindrance in the reaction of amine 3e with chloroacetyl chloride. The isolated yields of the corresponding CMP derivatives 13b–d are much lower, mainly due to their higher polarity and as a consequence more difficult chromatographic purification.
The CMPO ligands 9b–e could be identified based on the characteristic multiplets for the aromatic hydrogens at about 7.45 (18 H) and 7.85 ppm (12 H) and CMP ligands 13b–d based on a multiplet for the ethoxy group at around 4.15 ppm (12 H).
In general, the yields of the trialkylbenzene-based CMP(O) ligands 10a,b, 11, 12, and 14a,b are lower than those of the corresponding C-pivot-based ligands. In the case of the secondary amide containing ligands 10a,b and 14a,b (R2 = H) the 1H NMR spectra exhibit a doublet for the CH2P(O) methylene bridge (coupling with phosphorus) at ∼3.31 and ∼2.85 ppm, respectively. This probably indicates the conformational preference for the trans form of the secondary amide bonds. Only in the case of CMPO ligand 11 the 1H NMR spectrum shows a doublet for the CH2P(O) methylene bridge, indicating the existence of a dominating conformer of this molecule.
To investigate the influence on the complexation behavior of two different types of ligating sites on the C-pivot platform, tripodal amine 3d was treated with chloroacetyl chloride followed by reaction with a 1 ∶ 1 mixture of ethyl diphenyl phosphinite and triethyl phosphite. From the reaction mixture ligand 15, containing two CMPO arms and one CMP, was isolated in 11% yield. The 1H NMR spectrum displays the characteristic signals for the CMPO and CMP moieties in a 2 ∶ 1 ratio (Scheme 5).
 | ||
| Scheme 5 | ||
Malonamide methyl ester 16, obtained by reaction of methyl 3-chloro-3-oxopropionate and N-methyl-N-butylamine, was directly introduced on the C-pivot amine platforms 3a–d resulting in ligands 17a–d, and on the trialkylphenyl amines 7a,b giving ligands 18a,b (Scheme 6).
 | ||
| Scheme 6 | ||
The 1H NMR spectra of the malonamide ligands 17a and 18a,b, having one secondary and one tertiary amide moiety, show for the NCH3 group two singlets at about 3 ppm. The more rigid ligands 17b–d exist as a mixture of conformers giving rise to complicated 1H NMR spectra. All ligands exhibit the correct molecular ion peak in the HR-FAB mass spectra.
Extraction
In order to study whether CMP(O) ligands 9–15 and malonamides 17 and 18 are good ionophores for actinide/lanthanide separation, spike extraction tests were performed with Am3+ and Am3+ as model cations. The extractions were carried out with 1,1,2,2-tetrachloroethane (TCE) and n-octanol as the organic phase at two nitric acid concentrations.18From Table 1 and Fig. 1 it is clear that in the case of the C-pivot CMPO ligands 9b–d there is a positive influence of the N-alkyl substituents on the D (distribution) coefficients for both cations, solvents, and acidities. With CMPO ligand 9b in n-octanol there is an increase in D of 90 times compared with 9a. Apparently, in 9e the N-substituent is too crowded, since there is hardly any effect on the D coefficient. In the cases of 9c,d it is assumed that the lipophilic alkyl chains enhance the solubility of the complexes and consequently give rise to a better extraction efficiency. For all ligands 9a–e the separation factors are ∼2 in TCE and 2.5 in n-octanol, values that are also reported for other CMPO ligands.3,4
 | ||
| Fig. 1 Distribution and selectivity coefficients of CMPO ligands in 3 M HNO3–TCE (cL as in Table 1). | ||
| Ligand | Solvent and HNO3 concentration | |||||
|---|---|---|---|---|---|---|
| TCEb | n-Octanol | |||||
| Concentration/M | 1 M | 1 M | 3 M | |||
| a Data from ref. 9. b Data for 3 M HNO3 are presented in Fig. 1. | ||||||
| 9a a | 1.4 × 10–2 | Am | 0.51 | Am | 6.9 × 10–3 | 2.1 × 10–2 |
| Eu | 0.26 | Eu | 3.6 × 10–3 | 1.3 × 10–2 | ||
| 9b | 4.9 × 10–2 | Am | 1.4 | Am | 0.21 | 1.7 |
| Eu | 0.68 | Eu | 0.09 | 0.71 | ||
| 9c | 4.4 × 10–2 | Am | 1.1 | Am | 0.14 | 0.87 |
| Eu | 0.50 | Eu | 0.06 | 0.41 | ||
| 9d | 4.6 × 10–2 | Am | 3.2 | Am | 0.42 | 3.7 |
| Eu | 1.5 | Eu | 0.17 | 1.5 | ||
| 9e | 3.2 × 10–2 | Am | 0.18 | Am | 2.9 × 10–2 | 0.13 |
| Eu | 8.7 × 10–2 | Eu | 2.0 × 10–2 | 7.5 × 10–2 | ||
| 10a | 3.5 × 10–2 | Am | 2.0 | Am | Not soluble | |
| Eu | 1.2 | Eu | ||||
| 10b | 3.1 × 10–2 | Am | 1.0 | Am | 1.28 | 1.32 |
| Eu | 0.7 | Eu | 0.66 | 0.93 | ||
| 11 | 3.0 × 10–2 | Am | 0.02 | Am | 4.8 × 10–3 | 0.02 |
| Eu | 0.01 | Eu | 6.3 × 10–3 | 0.02 | ||
| 12 | 3.2 × 10–2 | Am | 0.16 | Am | 0.21 | 0.05 |
| Eu | 0.07 | Eu | 0.11 | 0.03 | ||
The trialkylbenzene platform-based CMPO ligands 10a,b have larger D coefficients (up to ∼200 times higher for 10b in n-octanol) than the corresponding C-pivot analogue 9a. Apparently, the preorganization on the same face of the molecule is beneficial. Unfortunately, there is hardly any Am3+/Am3+ selectivity. In this series N-alkylation with 3-pentyl chains gives rise to much smaller D coefficients. This is probably caused by the enlarged stiffness of the chelating groups, which is not compensated by a flexible spacer (here only 1 or 2 C atoms) between the platform and the CMPO group. A similar effect has been observed for calix[4]arene-based CMPO ligands3e with the ligating arms directly introduced at the benzene rings of the calix[4]arene.
In general, the CMP ligands are less effective than the corresponding CMPO derivatives (Table 2). The extraction behavior of the C-pivot CMP ligands 13b–d shows more or less the same trend as the corresponding CMPO ligands 9b–d upon substitution of the N-atom with different alkyl chains. In this series CMP ligands 13b and 13d are the most efficient at 3 M HNO3 in TCE. Attachment of three CMP chelating groups to a trialkylbenzene platform (14a,b) in general gave rise to rather low D coefficients.
| Ligand | Solvent and HNO3 concentration | ||||||
|---|---|---|---|---|---|---|---|
| TCE | n-Octanol | ||||||
| Concentration/M | 1 M | 3 M | 1 M | 3 M | |||
| a Data from ref. 9. | |||||||
| 13a a | 1.5 × 10–2 | Am | Precipitate | Precipitate | Am | <10–3 | 1.0 × 10–2 |
| Eu | Precipitate | Precipitate | Eu | <10–3 | 9.9 × 10–3 | ||
| 13b | 7.8 × 10–2 | Am | 1.5 × 10–2 | 0.3 | Am | 8.6 × 10–3 | 4.0 × 10–2 |
| Eu | 1.4 × 10–2 | 0.2 | Eu | 5.9 × 10–3 | 2.8 × 10–2 | ||
| 13c | 5.1 × 10–2 | Am | 7.5 × 10–3 | 0.1 | Am | 4.6 × 10–3 | 2.4 × 10–2 |
| Eu | 4.9 × 10–3 | 0.12 | Eu | 3.3 × 10–3 | 1.8 × 10–2 | ||
| 13d | 5.5 × 10–2 | Am | 1.8 × 10–2 | 0.42 | Am | 6.3 × 10–3 | 3.3 × 10–2 |
| Eu | 2.0 × 10–2 | 0.54 | Eu | 4.4 × 10–3 | 2.5 × 10–2 | ||
| 14a | 4.5 × 10–2 | Am | <10–3 | 2.8 × 10–2 | Am | 3.2 × 10–3 | 2.0 × 10–2 |
| Eu | <10–3 | 5.2 × 10–2 | Eu | 4.1 × 10–3 | 4.8 × 10–2 | ||
| 14b | 7.2 × 10–2 | Am | 4.0 × 10–3 | 0.11 | Am | 1.8 × 10–2 | 3.6 × 10–2 |
| Eu | 3.3 × 10–3 | 8.1 × 10–2 | Eu | 1.3 × 10–2 | 2.5 × 10–2 | ||
| 15 | 3.2 × 10–2 | Am | 0.67 | 5.0 | Am | 0.03 | 0.26 |
| Eu | 0.39 | 3.5 | Eu | 0.02 | 0.19 | ||
Mixed ligand 15, containing two CMPO and one CMP moiety does not show improved extraction properties. Its D coefficients are even slightly lower than the averaged value of those of the corresponding CMPO ligand 9d and CMP ligand 13d.
Tripodal malonamides 17a–d and 18a,b show low distribution values similar to those for the simple non-preorganized malonamides.7,12d In the case of the C-pivot ligands 17a–dN-alkylation has a negative effect on the extraction behavior, the malonamides 17b–d hardly extract (Table 3). Possibly six secondary amide groups generate a highly rigid structure and as a result the ligand is not able to fold properly around a metal ion. For the ligands 17a and 18a,b the D coefficients are higher in n-octanol than in TCE. Apparently, intramolecular H-bonding is disfavored in the polar n-octanol resulting in a better extraction efficiency.
| Ligand | Solvent and HNO3 concentration | ||||||
|---|---|---|---|---|---|---|---|
| TCE | n-Octanol | ||||||
| Concentration/M | 1 M | 3 M | 1 M | 3 M | |||
| 17a | 9.9 × 10–2 | Am | 1.1 × 10–2 | 0.18 | Am | 0.17 | 0.47 |
| Eu | 8.5 × 10–3 | 0.16 | Eu | 0.13 | 0.41 | ||
| 17b | 6.2 × 10–2 | Am | <10–3 | 5.6 × 10–3 | Am | 4.6 × 10–3 | 1.6 × 10–2 |
| Eu | <10–3 | 5.6 × 10–3 | Eu | 3.3 × 10–3 | 1.3 × 10–2 | ||
| 17c | 4.6 × 10–2 | Am | <10–3 | 3.1 × 10–3 | Am | 3.4 × 10–3 | 1.2 × 10–2 |
| Eu | <10–3 | 4.2 × 10–3 | Eu | 2.2 × 10–3 | 1.1 × 10–2 | ||
| 17d | 6.0 × 10–2 | Am | 1.3 × 10–3 | 8.9 × 10–3 | Am | 1.7 × 10–3 | 7.6 × 10–3 |
| Eu | 1.2 × 10–3 | 8.9 × 10–3 | Eu | 1.1 × 10–3 | 6.7 × 10–3 | ||
| 18a | 5.5 × 10–2 | Am | <10–3 | 2.0 × 10–2 | Am | 0.12 | 0.26 |
| Eu | <10–3 | 5.3 × 10–2 | Eu | 9.1 × 10–2 | 0.26 | ||
| 18b | 4.3 × 10–2 | Am | <10–3 | 9.5 × 10–3 | Am | 0.12 | 0.25 |
| Eu | <10–3 | 1.8 × 10–2 | Eu | 0.11 | 0.23 | ||
Using C-pivot CMPO ligands 9b,d the extraction rate was studied. In both cases the extraction is a fast process with the equilibrium reached in less than 5 min in micro-tubes shaken with a vortex device. For all ligands the distribution coefficients are higher at 3 M than at 1 M nitric acid. This allows the use of diluted HNO3 to reverse the extraction process and to “strip” the organic phase from the metal ions, and the recovery of the ligand. In the case of ligands 9b,d stripping of the complexes was performed with 0.1 M HNO3 in n-octanol within 5 min.
Potentiometric measurements
Selected C-pivot tripodal CMPO (9b–d), CMP (13b–d), and malonamide (17a–d) ligands were examined for their complexing ability using the potentiometric so-called “sandwich method” and in polymeric membrane ion-selective electrodes. Stability constants and selectivity coefficients for Na+, K+, Mg2+, Ca2+, Cu2+, Pb2+ or UO22+ are summarized in Tables 4 and 5, respectively.| Ligand | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CMPO | CMP | Malonamides | ||||||||||
| Cation | 9a b | 9b | 9c | 9d | 13a b | 13b | 13c | 13d | 17a | 17b | 17c | 17d |
| a Standard deviations <0.3 (from at least three replicate measurements). b Data from ref. 9. | ||||||||||||
| Na+ | 8.4 | 8.4 | 8.4 | 8.5 | 4.8 | 5.4 | 8.1 | 7.5 | 3.6 | 6.5 | 6.6 | 6.9 |
| K+ | — | 7.4 | 6.0 | 6.1 | — | 4.2 | 6.0 | 5.3 | 2.0 | 4.9 | 4.9 | 4.5 |
| Mg2+ | 16.4 | 19.8 | 19.8 | 19.0 | — | 13.3 | 16.2 | 16.2 | 10.6 | 15.5 | 15.8 | 16.6 |
| Ca2+ | 17 | 20.5 | 20.9 | 21.5 | — | 14.0 | 18.3 | 16.8 | 8.5 | 15.8 | 15.8 | 16.5 |
| Cu2+ | 19.1 | 20.5 | 20.0 | 20.1 | 11.8 | 13.0 | 16.3 | 15.0 | 11.7 | 17.2 | 17.3 | 16.8 |
| Pb2+ | 17.4 | 21.3 | 20.7 | 21.5 | 9.0 | 14.8 | 18.1 | 16.3 | 10.5 | 16.7 | 16.8 | 16.6 |
| UO22+ | 21.5 | 23.1 | 23.1 | 23.1 | 12.3 | 15.9 | 19.2 | 17.4 | 12.5 | 19.4 | 19.3 | 19.5 |
| Ligand | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CMPO | CMP | Malonamides | ||||||||||
| Cation | 9a b | 9b | 9c | 9d | 13a b | 13b | 13c | 13d | 17a | 17b | 17c | 17d |
| a Standard deviations <0.3 (from at least three replicate measurements). b Data from ref. 9. | ||||||||||||
| Na+ | –4.6 | –5.8 | –5.7 | –6.0 | –2.3 | –3.5 | –3.1 | –3.6 | –3.0 | –3.7 | –3.8 | –3.0 |
| K+ | — | –6.6 | –6.7 | –6.4 | — | –5.9 | –4.1 | –5.1 | –1.8 | –4.1 | –3.8 | –3.8 |
| Mg2+ | — | –2.5 | –2.6 | –2.3 | — | –2.3 | –2.2 | –2.2 | –0.8 | –2.3 | –2.5 | –2.5 |
| Ca2+ | — | –1.4 | –1.1 | –1.0 | — | –1.2 | –0.9 | –0.7 | –1.2 | –1.4 | –1.4 | –1.4 |
| Cu2+ | –0.9 | –2.0 | –2.0 | –1.3 | –1.3 | –2.3 | –2.7 | –2.3 | –0.5 | –0.5 | –0.7 | –0.8 |
| Pb2+ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| UO22+ | 4.1 | 2.5 | 2.7 | 2.2 | 2.4 | 1.0 | 1.0 | 1.3 | 1.9 | 2.8 | 2.8 | 2.7 |
To simplify the discussion and to show the influence of functional groups (donor atoms) on the complexing capability of the examined ligands, three N-substituted compounds with an octyl chain were selected (9c, 13c, and 17c).
From Table 4 it is clear that all the examined ligands form the strongest complexes with the uranyl cation. This behavior can be explained by Pearson’s soft–hard acid and base theory. All ligands possess hard oxygen atoms in the ligating centers that assures a strong interaction with the hard UO22+ cation.
Ligand 9c, representing the CMPO series, exhibits the highest complexing ability towards all studied cations. Just as for the D coefficients for Am3+ and Am3+, the complex formation constants are higher for ligands with phenyl groups (CMPO) than for those possessing ethoxy moieties (CMP). Ligand 17c, as a representative of the malonamide group, gave the weakest complexes. However, it exhibits a higher preference toward Cu2+ (log βILn = 17.3) than CMP ligand 13c (log βILn = 16.3). It is well known that metal cations interact more strongly with ligands, that, besides P, O, N atoms, also possess aromatic rings. The strong complexation properties of 9c can be related to the presence of six aromatic rings and possibly some metal–ligand aromatic interactions (MLACπ).19
The CMPO (9c), CMP (13c), and malonamide (17c) derivatives were examined as potential ligands in 2-nitrophenyl octyl ether (o-NPOE)–PVC membranes in terms of electrode selectivities. The logarithmic values of the selectivity coefficients calculated for Pb2+ as the primary cation are presented in Fig. 2. It is well known that selectivity coefficients for neutral carrier-based membranes are mainly related to the stability constants of ion–ionophore complexes in the membrane.20
![Selectivity coefficients for electrodes with membranes based on compounds 9c, 13c, and 17c and with only tetrakis[3,4-bis(trifluoromethyl)phenyl]borate (KTFPB).](/image/article/2007/NJ/b613254e/b613254e-f2.gif) | ||
| Fig. 2 Selectivity coefficients for electrodes with membranes based on compounds 9c, 13c, and 17c and with only tetrakis[3,4-bis(trifluoromethyl)phenyl]borate (KTFPB). | ||
The results reveal that the ionophores 9c, 13c, and 17c induce a selectivity that differs from the so-called Hofmeister series.21,22 As expected based on the stability constants (Table 4), the electrodes show an enhanced selectivity towards UO22+. In the case of compound 9c the interaction with the uranyl cation is so strong (log βILn = 23.1) that the electrode had an upper detection limit (c = 10–3 M). Above this concentration flattening of the calibration curve occurs, which can be explained by coextraction of anions from the aqueous sample solution. However, in the 10–5–10–3 M concentration range a linear response with a Nernstian slope can be observed. Since compounds 13c and 17c form weaker complexes with UO22+, the corresponding electrodes give a linear response in the whole UO22+ concentration range studied (Fig. 3).
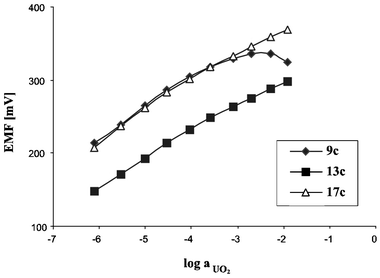 | ||
| Fig. 3 Calibration curves of electrodes doped with compounds 9c, 13c, and 17c towards UO22+. | ||
The incorporation of these ligands into a polymeric membrane leads to considerable changes in the selectivity for Pb2+ over K+ and Na+. It suggests a very weak interaction between the ligands and K+, Na+ in comparison to the electrodes containing a membrane with an ion-exchanger (KTFPB). The smallest selectivity changes are observed for Ca2+ and Cu2+, which can be attributed to the comparable stability constants of the Pb2+, Ca2+, and Cu2+ complexes (Tables 4 and 5).
It should be pointed out that membranes doped with CMPO ligand 9c exhibit a better differentiation of the selectivity coefficients than those doped with CMP ligand 13c (logKpotPb,UO2 ≈ 3 and logKpotPb,Na ≈ –6 for 9c; logKpotPb,UO2 ≈ 1 and logKpotPb,Na ≈ –3 for 13c). Taking into account the low K+ and Na+ selectivity coefficients in the case of the electrode with CMPO 9c doped membrane, this electrode may be very useful for the determination of UO22+ in the presence of high levels of alkali metal cations.
Beside the influence of the different types of ligands (CMPO, CMP, malonamide) on the complex stability constants and the selectivity coefficients, the influence of minor structural changes within the ligand groups was also studied. In all cases ligands substituted with alkyl chains (9b–d, 13b–d, and 17b–c) form stronger complexes with the examined cations than those having an H atom in the same position (9a, 13a, 17a, respectively). However, the length and the structure of an alkyl chain only have a slight influence on the strength of the interaction between ligands and metal cations. Whereas CMPO and CMP ligands in general maintain the same behavior within the series of compounds for both extraction and potentiometric studies, the malonamides behave differently. In extraction experiments the N-substituted malonamides 17b–d revealed almost no extraction abilities, while in the potentiometric measurements they formed more stable complexes than 17a.
Where the selectivity coefficients in the malonamide series are concerned, ionophore 17a does not discriminate against K+ so much as the other ions (Fig. 4). In line with the influence of the alkyl chains on the stability constants in the case of the N-substituted malonamides 17b–d the selectivity coefficients only show small differences.
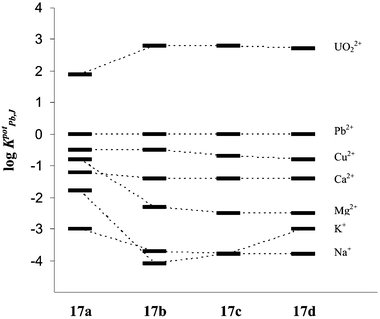 | ||
| Fig. 4 Selectivity coefficients for electrodes with membranes based on compounds 17a–d. | ||
From Table 4 it is clear that in the CMPO ligand series neither length nor shape of the alkyl substituent have any influence on the stability constants. In the CMP ligand series the attachment of a long linear alkyl chain (13c) leads to larger stability constants compared to 13b or 13d, having a short and branched alkyl chain, respectively. The trend is opposite to that of the extraction results.
There are hardly differences between the selectivity coefficients of the short (C4 and C5) and long (C8) alkyl-substituted CMPO and CMP ligands (Table 5). Despite the differences in the stability constants between the alkyl-substituted CMP ligands 13b–d, the selectivity coefficients of electrodes doped with these ligands are almost identical. The reason for this behavior is that the relative differences between the stability constants of the complexes of ligands 13c with the different metal cations (for instance log βILn = 16.3 for Cu2+ and log βILn = 19.2 for UO22+, Δ log βILn = 2.9) are very similar to the differences in the stability constants of the corresponding complexes of ligand 13b (log βILn = 13.0 for Cu2+ and log βILn = 15.9 for UO22+) or 13d (log βILn = 14.9 for Cu2+ and log βILn = 17.4 for UO22+) with the same cations.
Conclusions
Series of tripodal ligands based on the C-pivot and trialkylbenzene platforms bearing (N-substituted) CMP(O) and malonamide moieties were prepared. In general, the C-pivot CMP(O) ligands 9a–e and 13a–d show a positive effect of the N-alkyl substituent on the D coefficients for Am3+ and Am3+ extraction. The trialkylbenzene CMPO ligands 10a,b have considerably larger D coefficients than the corresponding C-pivot analogues, probably due to preorganization of the ligating sites on one face of the platform. In this series N-alkylation with 3-pentyl chains negatively influences the D coefficients probably due to the enlarged stiffness of the ligating sites which is not compensated for by a flexible spacer between the platform and the CMPO moieties. For the malonamides 17a–dN-alkylation has a negative effect on the extraction behavior, possibly because 17b–d are too rigid to properly fold around a metal cation.In general, there is good coherence between the extraction data and those of the potentiometric measurements, except in the case of the N-substituted malonamides.
Experimental
Synthesis
1H and 13C NMR spectra were recorded on a Varian Unity INOVA 300 MHz and a Varian Unity 400 WB NMR spectrometer, respectively. Residual solvent protons were used as an internal standard and chemical shifts are given in ppm relative to tetramethylsilane (TMS). Fast atom bombardment (FAB) mass spectra were measured on a Finnigan MAT 90 spectrometer using m-nitrobenzyl alcohol (NBA) as a matrix. Elemental analyses were carried out using a 1106 Carlo-Erba Strumentazione element analyser. All solvents were purified by standard procedures. All other chemicals were analytically pure and were used without further purification. Melting points (uncorrected) of all compounds were obtained on a Reichert melting point apparatus.C-Pivot tripodal amine 3b. Reaction of nitrile 1 (2.24 g, 7.64 mmol), amine 2b (50 mL, 505 mmol) and 10% Pd–C (1.43 g) as a catalyst gave 3b as a pale yellow oil (3.57 g, 99%). δH(300 MHz; CDCl3) 3.44 (6 H, t, J 6.2, OCH2CH2CH2N), 3.25 (6 H, s, CCH2O), 2.69 (6 H, t, J 6.7, OCH2CH2CH2N), 2.60 (6 H, t, J 7.4, NCH2CH2CH2CH3), 1.74 (6 H, quintet, J 6.7 OCH2CH2CH2N), 1.28–1.62 (17 H, m, NCH2CH2CH2CH3, CCH2CH3, NH), 0.91 (9 H, t, J 7.2, NCH2CH2CH2CH3), 0.83 (3 H, t, J 7.5, CCH2CH3); δC(75 MHz; CDCl3) 71.7, 70.4, 50.1, 48.0, 43.3, 32.6, 30.4, 23.5, 20.8, 14.2, 7.9; m/z (FAB) 474.4597 ([M + H]+ C27H60N3O3 requires 474.4635).
C-Pivot tripodal amine 3c. Reaction of nitrile 1 (1.76 g, 5.99 mmol), amine 2c (40 mL, 242 mmol) and 10% Pd–C (1.16 g) as a catalyst gave 3c as an oil (3.72 g, 97%). δH(300 MHz; CDCl3) 3.44 (6 H, t, J 6.2, OCH2CH2CH2N), 3.25 (6 H, s, CCH2O), 2.69 (6 H, t, J 6.9, OCH2CH2CH2N), 2.59 (6 H, t, J 7.1, NCH2C6H12CH3), 1.74 (6 H, quintet, J 6.5, OCH2CH2CH2N), 1.20–1.55 (41 H, m, NCH2C6H12CH3, CCH2CH3, NH), 0.81–0.91 (12 H, m, NCH2C6H12CH3, CCH2CH3); δC(75 MHz; CDCl3) 71.7, 70.4, 50.4, 48.0, 43.3, 32.0, 30.5, 30.3, 29.8, 29.5, 27.7, 23.5, 22.8, 14.2, 7.9; m/z (FAB) 642.6481 ([M + H]+ C39H84N3O3 requires 642.6513).
C-Pivot tripodal amine 3d. Reaction of nitrile 1 (2.11 g, 7.19 mmol), amine 2d (25 g, 291 mmol) and 10% Pd–C (0.99 g) as a catalyst gave 3d as an oil (3.59 g, 97%). δH(300 MHz; CDCl3) 3.44 (6 H, t, J 6.0, OCH2CH2CH2N), 3.26 (6 H, s, CCH2O), 2.66 (6 H, t, J 7.0, OCH2CH2CH2N), 2.37 (3 H, quintet, J 6.3, NCH(CH2CH3)2), 1.73 (6 H, quintet, J 6.5, OCH2CH2CH2N), 1.35–1.48 (14 H, m, CH2CH3, NCH(CH2CH3)2), 0.88 (18 H, t, J 7.5, NCH(CH2CH3)2), 0.83 (3 H, t, J 7.8, CH2CH3); δC(75 MHz; CDCl3) 71.8, 70.4, 60.3, 44.8, 43.3, 30.7, 26.1, 23.5, 10.1, 8.0; m/z (FAB) 516.5127 ([M + H]+ C30H66N3O3 requires 516.5104).
C-Pivot tripodal amine 3e. Reaction of nitrile 1 (1.33 g, 4.54 mmol), amine 2e (30 mL, 256 mmol) and 10% Pd–C (0.82 g) as a catalyst gave 3e as an oil (2.07 g, 88%). δH(300 MHz; CDCl3) 3.42 (6 H, t, J 6.0, OCH2CH2CH2N), 3.25 (6 H, s, CCH2O), 2.56 (6 H, t, J 6.7, OCH2CH2CH2N), 1.68 (6 H, quintet, J 6.0, OCH2CH2CH2N), 1.39 (8 H, q, J 7.4, CH2CH3), 1.01 (18 H, s, N(C(CH3)2), 0.83 (12 H, t, J 7.4, CH2CH3); δC(75 MHz; CDCl3) 71.8, 70.5, 52.5, 43.3, 39.8, 33.3, 31.3, 26.8, 23.5, 8.5, 8.0; m/z (FAB) 516.5111 ([M + H]+ C30H67N3O3 requires 516.5104).
CMPO ligand 9b. Reaction of tripodal amine 3b (1.25 g, 2.63 mmol), chloroacetyl chloride (1.9 mL, 23.9 mmol) and K2CO3 (3.5 g, 25 mmol) and subsequently diphenyl ethyl phosphinite (2.3 mL, 10.6 mmol) gave ligand 9b (2.72 g, 86%). δH(300 MHz; CDCl3) 7.80–7.88 (12 H, m, Ph), 7.39–7.53 (18 H, m, Ph), 3.07–3.60 (30 H, m, CH2O, CH2N, CH2P(O)), 1.64–1.76, 1.42–1.58, 1.08–1.35 (20 H, m, CH3CH2C, CH3CH2CH2CH2N, OCH2CH2CH2N), 0.65–0.92 (12 H, m, CH3); δC(75 MHz; CDCl3) 165.1, 133.5, 133.4, 132.2, 131.5, 131.4, 128.8, 128.6, 71.6, 68.9, 68.0, 49.4, 46.2, 46.0, 44.2, 43.2, 38.9, 38.6, 38.0, 37.7, 31.3, 29.8, 29.2, 28.1, 23.5, 20.3, 14.1, 8.0; m/z (FAB) 1200.6077 ([M + H]+ C69H93N3O9P3 requires 1200.6125).
CMPO ligand 9c. Reaction of tripodal amine 3c (0.88 g, 1.37 mmol), chloroacetyl chloride (0.9 mL, 11.4 mmol) and K2CO3 (1.8 g, 13 mmol) and subsequently diphenyl ethyl phosphinite (2.0 mL, 8.7 mmol) gave ligand 9c (1.66 g, 88%). δH(300 MHz; CDCl3) 7.80–7.87 (12 H, m, Ph), 7.38–7.54 (18 H, m, Ph), 3.05–3.59 (30 H, m, CH2O, CH2N, CH2P(O)), 1.64–1.74, 1.42–1.56, 1.08–1.32 (44 H, m, CH3CH2C, CH3C6H12CH2N, OCH2CH2CH2N), 0.85 (9 H, t, J 6.7, CH3C7H14N) 0.64–0.78 (3 H, m, CH3CH2C); δC(75 MHz; CDCl3) 165.0, 133.5, 133.4, 132.2, 131.5, 131.3, 128.8, 128.6, 71.7, 68.9, 68.0, 49.7, 46.4, 46.0, 44.1, 43.2, 38.5, 38.0, 32.0, 31.9, 29.6, 29.4, 29.2, 28.1, 27.7, 27.1, 27.0, 22.8, 22.7, 14.2, 8.0; m/z (FAB) 1406.7613 ([M + K]+ C81H116N3O9P3K requires 1406.7562).
CMPO ligand 9d. Reaction of tripodal amine 3d (0.93 g, 1.80 mmol), chloroacetyl chloride (1.3 mL, 16.2 mmol) and K2CO3 (2.8 g, 20 mmol) and subsequently diphenyl ethyl phosphinite (2.5 mL, 11.6 mmol) gave ligand 9d (1.87 g, 83%). δH(300 MHz; CDCl3) 7.76–7.89 (12 H, m, Ph), 7.39–7.47 (18 H, m, Ph), 3.98–4.08, 3.56–3.74, 3.03–3.42 (27 H, m, CH2O, CH2NCH, CH2P(O)), 1.72–1.84, 1.18–1.62 (20 H, m, CH3CH2C, NCH(CH2CH3)2, OCH2CH2CH2N), 0.62–0.90 (21 H, m, CH3); δC(75 MHz; CDCl3) 166.2, 166.1, 166.0, 165.9, 133.8, 133.6, 132.4, 132.2, 132.1, 131.5, 131.4, 128.7, 128.6, 71.6, 69.7, 68.7, 62.3, 58.8, 53.6, 43.3, 42.1, 39.6, 38.5, 38.2, 31.4, 29.1, 26.5, 26.0, 23.5, 11.3, 8.0; m/z (FAB) 1280.6150 ([M + K]+ C72H98N3O9P3K requires 1280.6153).
CMPO ligand 9e. Reaction of tripodal amine 3e (0.86 g, 1.67 mmol), chloroacetyl chloride (1.2 mL, 15 mmol) and K2CO3 (2.2 g, 16 mmol) and subsequently diphenyl ethyl phosphinite (1.5 mL, 6.9 mmol) gave ligand 9e (0.27 g, 13%). δH(300 MHz; CDCl3) 7.81–7.86 (12 H, m, Ph), 7.40–7.52 (18 H, m, Ph), 3.65 (6 H, d, JP–H 15.3, CH2P(O)), 3.20–3.60 (12 H, m, OCH2CH2CH2N), 3.06 (6 H, s, CCH2O), 2.20–2.32 (6 H, m, OCH2CH2CH2N), 1.68–1.71 (8 H, m, CH2CH3), 1.26 (18 H, s, CCH3), 0.65 (12 H, t, J 7.4, CH2CH3); δC(75 MHz; CDCl3) 165.7, 165.6, 133.8, 132.5, 131.9, 131.5, 131.3, 128.7, 128.5, 71.7, 68.6, 61.4, 44.2, 43.2, 41.0, 40.2, 32.5, 32.1, 27.0, 23.2, 8.9, 8.0; m/z (FAB) 1280.5921 ([M + K]+ C72H98N3O9P3K requires 1280.6153).
CMPO ligand 10a. Reaction of tripodal amine 7a (0.67 g, 2.69 mmol), chloroacetyl chloride (2.03 mL, 25.6 mmol) and K2CO3 (3.6 g, 26 mmol) and subsequently diphenyl ethyl phosphinite (2 mL, 9.3 mmol) gave ligand 10a (1.18 g, 45%) (Found: C, 68.29; H, 5.99; N, 3.80. Calc. for C57H60N3O6P3·0.33CH2Cl2: C, 68.58; H, 6.09; N, 4.18%); mp > 221 °C (melting with decomposition); δH(300 MHz; CDCl3) 7.71–7.78, 7.42–7.58 (33 H, m, NH, Ph), 3.31 (6 H, d, JH–P 12.3, C(O)CH2P(O)), 3.15–3.23, 2.64–2.70 (12 H, m, PhCH2CH2N), 2.21 (9 H, s, CH3); δC(75 MHz; CDCl3) 164.91, 164.85, 133.9, 133.6, 132.7, 132.6, 132.5, 131.1, 131.0, 130.9, 129.2, 129.0, 39.4, 39.3, 38.6, 30.9, 16.0.
CMPO ligand 10b. Reaction of tripodal amine 7b (0.76 g, 2.62 mmol), chloroacetyl chloride (1.5 mL, 18.3 mmol) and K2CO3 (2.6 g, 19 mmol) and subsequently diphenyl ethyl phosphinite (2.3 mL, 10.6 mmol) gave ligand 10b (0.96 g, 36%) (Found: C, 70.05; H, 6.40; N, 4.23. Calc. for C60H66N3O6P3·0.1CH2Cl2: C, 70.08; H, 6.48; N, 4.08%); mp 143 °C; δH(300 MHz; CDCl3) 7.72–7.79, 7.42–7.57 (33 H, m, CH2NHCO, Ph), 3.32 (6 H, d, JH–P 12.3, C(O)CH2P(O)), 3.15–3.26, 2.52–2.64 (18 H, m, PhCH2CH2N CH2CH3), 1.02 (9 H, t, J 9.2, CH2CH3); δC(100 MHz; CDCl3) 164.81, 164.77, 140.5, 132.7, 132.6, 132.4, 132.2, 131.1, 131.0, 130.9, 129.2, 129.0, 41.0, 39.4, 38.8, 29.5, 22.5, 15.9.
CMPO ligand 11. Reaction of tripodal amine 8 (0.72 g, 1.44 mmol), chloroacetyl chloride (1.0 mL, 12.6 mmol) and K2CO3 (1.8 g, 13 mmol) and subsequently diphenyl ethyl phosphinite (4.57 mL, 21.1 mmol) gave ligand 11 (1.22 g, 69%). δH(300 MHz; CDCl3) 7.84–7.96 (12 H, m, Ph), 7.42–7.54 (18 H, m, Ph), 3.80–3.91 (3 H, m, NCH(CH2CH3)2), 3.70 (6 H, d, JP–H 15.6, CH2P(O)), 3.01–3.10 (6 H, m, NCH2CH2Ph), 2.81 (6 H, q, J 7.8, CH3CH2Ph), 2.37–2.46 (6 H, m, NCH2CH2Ph), 1.42–1.70 (12 H, m, NCH(CH2CH3)2), 0.79–1.00 (27 H, m, CH3); δC(75 MHz; CDCl3) 166.4, 166.3, 141.3, 133.3, 133.2, 132.2, 131.9, 131.6, 131.4, 128.8, 128.6, 62.4, 42.8, 40.1, 39.2, 27.9, 26.6, 22.6, 15.7, 11.3; m/z (FAB) 1266.6274 ([M + K]+ C75H96N3O6P3K requires 1266.6149).
CMPO ligand 12. Reaction of tripodal amine 5 (0.62 g, 1.34 mmol), chloroacetyl chloride (0.9 mL, 11.3 mmol) and K2CO3 (1.8 g, 13 mmol) and subsequently diphenyl ethyl phosphinite (2 mL, 9.26 mmol) gave ligand 12 (0.51 g, 32%). δH(300 MHz; CDCl3) 7.85–7.92 (12 H, m, Ph), 7.44–7.57 (18 H, m, Ph), 4.45–4.67 (6 H, m, PhCH2N), 3.66–3.80 (6 H, m, C(O)CH2P(O)), 2.35–2.73 (9 H, m, NHCH(CH2CH3)2, PhCH2CH3), 1.42–1.62 (12 H, m, NHCH(CH2CH3)2), 0.40–1.25 (27 H, m, CH3); δC(75 MHz; CDCl3) 165.8, 146.5, 133.5, 132.3, 132.2, 131.4, 131.3, 131.2, 128.9, 128.8, 128.6, 61.7, 47.8, 40.9, 40.1, 24.0, 22.8, 16.1, 11.6, 11.5; m/z (FAB) 1224.5672 ([M + K]+ C72H90N3O6P3K requires 1224.5680).
CMP ligand 13b. Reaction of tripodal amine 3b (0.91 g, 1.91 mmol), chloroacetyl chloride (1.2 mL, 15.3 mmol) and K2CO3 (2.2 g, 16 mmol) and subsequently triethyl phosphite (4.0 mL, 24.1 mmol) gave ligand 13b (1.31 g, 68%). δH(300 MHz; CDCl3), 4.11–4.21 (12 H, m, POCH2CH3), 3.24–3.48 (24 H, m, CH2O, CH2N), 2.97–3.09 (6 H, m, C(O)CH2P(O)), 1.72–1.85, 1.25–1.61 (38 H, m, CH3CH2C, CH3CH2CH2CH2N, OCH2CH2CH2N, P(O)CH2CH3), 0.80–0.97 (12 H, m, CH3CH2C, CH3CH2CH2CH2N); δC(100 MHz; CDCl3) 164.7, 164.6, 164.5, 71.7, 68.9, 67.9, 62.7, 62.6, 49.3, 46.0, 45.8, 44.1, 43.2, 34.2, 34.0, 32.8, 32.7, 31.3, 29.8, 29.3, 28.1, 23.4, 20.3, 20.2, 16.6, 16.5, 14.0, 8.0; m/z (FAB) 1008.5818 ([M + H]+ C45H93N3O15P3 requires 1008.5820).
CMP ligand 13c. Reaction of tripodal amine 3c (1.40 g, 2.18 mmol), chloroacetyl chloride (1.5 mL, 18.5 mmol) and K2CO3 (2.6 g, 19 mmol) and subsequently triethyl phosphite (5.0 mL, 29.2 mmol) gave ligand 13c (0.82 g, 32%). δH(300 MHz; CDCl3) 4.09–4.19 (12 H, m, POCH2CH3), 3.20–3.47 (24 H, m, CH2O, CH2N), 2.95–3.07 (6 H, m, C(O)CH2P(O)), 1.72–1.82, 1.17–1.58 (62 H, m, CH3CH2C, CH3C6H12CH2N, OCH2CH2CH2N, POCH2CH3) 0.79–0.88 (12 H, m, CH3CH2C, CH3C6H12CH2N); δC(75 MHz; CDCl3) 164.8, 164.6, 164.5, 71.6, 69.0, 68.0, 62.7, 62.6, 62.5, 49.6, 46.3, 45.8, 44.1, 43.3, 34.5, 32.7, 32.5, 32.0, 31.9, 29.6, 29.5, 29.4, 29.3, 28.2, 27.8, 27.2, 27.1, 23.5, 22.8, 16.6, 16.5, 14.2, 8.1; m/z (FAB) 1214.7168 ([M + K]+ C57H116N3O15P3K requires 1214.7256).
CMP ligand 13d. Reaction of tripodal amine 3d (0.87 g, 1.68 mmol), chloroacetyl chloride (1.2 mL, 15.1 mmol) and K2CO3 (2.2 g, 16 mmol) and subsequently triethyl phosphite (3.0 mL, 29.1 mmol) gave ligand 13d (0.37 g, 21%). δH(300 MHz; CDCl3) 4.09–4.19 (12 H, m, POCH2CH3), 3.00–3.08, 3.14–3.63 (27 H, m, CH2O, CH2PO, CH2NCH), 1.75–1.88, 1.35–1.59 (20 H, m, CH3CH2C, NCH(CH2CH3)2, OCH2CH2CH2N), 1.30 (16 H, t, J 3.5, POCH2CH3), 0.79–0.91 (21 H, m, CH3CH2C, NCH(CH2CH3)2); δC(75 MHz; CDCl3) 166.0, 165.9, 165.6, 165.5, 71.9, 71.7, 69.8, 68.7, 62.6, 62.5, 62.3, 58.6, 42.4, 41.9, 39.6, 35.0, 34.8, 33.1, 33.0, 31.6, 29.2, 26.5, 26.1, 23.5, 16.6, 16.5, 11.3, 8.0; m/z (FAB) 1088.5792 ([M + K]+ C48H98N3O15P3K requires 1088.5848).
CMP ligand 14a. Reaction of tripodal amine 7a (0.69 g, 2.78 mmol), chloroacetyl chloride (2.1 mL, 26.4 mmol) and K2CO3 (3.8 g, 28 mmol) and subsequently triethyl phosphite (6.0 mL, 34.9 mmol) gave ligand 14a (0.96 g, 44%). δH(400 MHz; CDCl3) 7.03 (3 H, t, J 5.8, CH2NHCO), 4.14 (12 H, m, OCH2CH2), 3.28–3.33, 2.86–2.92 (12 H, m, PhCH2CH2N), 2.85 (6 H, d, JH–P 20.4, C(O)CH2P(O)), 2.35 (9 H, s, PhCH3), 1.33 (18 H, t, J 6.8, OCH2CH3); δC(100 MHz; CDCl3) 164.24, 164.21, 134.0, 133.6, 62.9, 39.4, 35.9, 34.6, 30.9, 16.5, 16.1; m/z (FAB) 784.3481 ([M + H]+ C33H61N3O12P3 requires 784.3468).
CMP ligand 14b. Reaction of tripodal amine 7b (0.65 g, 2.24 mmol), chloroacetyl chloride (1.25 mL, 15.7 mmol) and K2CO3 (2.2 g, 16 mmol) and subsequently triethyl phosphite (6 mL, 35 mmol) gave ligand 14b (0.76 g, 41%). δH(400 MHz; CDCl3) 6.92 (3 H, t, J 5.8, CH2NHCO), 4.08–4.16 (12 H, m, OCH2CH2), 3.29–3.31, 2.77–2.81 (6 H, m, PhCH2CH2N), 2.82 (6 H, d, JH–P 20.4, C(O)CH2P(O)), 2.70 (6 H, q, J 7.2, PhCH2CH3), 1.31 (18 H, t, J 7.2, OCH2CH3), 1.11 (9 H, t, J 7.2, PhCH2CH3); δC(100 MHz; CDCl3) 164.2, 164.13, 140.7, 132.4, 63.0, 62.9, 41.1, 35.9, 34.6, 29.5, 22.7, 16.5, 16.1; m/z (FAB) 826.3972 ([M + H]+ C36H67N3O12P3 requires 826.3938).
CMPO/CMP ligand 15. Reaction of tripodal amine 3d (0.89 g, 1.73 mmol), chloroacetyl chloride (1.25 mL, 15.7 mmol) and K2CO3 (2.2 g, 16 mmol) and subsequently a mixture of triethyl phosphite (0.7 mL, 4.1 mmol) and diphenyl ethyl phosphinite (1 mL, 4.63 mmol) gave ligand 15 (0.22 g, 11%). δH(300 MHz; CDCl3) 7.80–7.89 (8 H, m, Ph), 7.40–7.49 (12 H, m, Ph), 4.12–4.18 (4 H, m, POCH2CH3), 3.98–4.08, 3.52–3.78, 3.01–3.42 (27 H, m, CH2O, CH2NCH, CH2P(O)), 1.66–1.86, 1.21–1.62 (26 H, m, CH3CH2C, NCH(CH2CH3)2, OCH2CH2CH2N, P(O)CH2CH3), 0.68–0.92 (21 H, m, CH3CH2C, NCH(CH2CH3)2); δC(75 MHz; CDCl3) 166.2, 166.02, 165.96, 165.6, 165.5, 133.6, 133.4, 132.2, 132.1, 131.5, 131.4, 128.73, 128.71, 128.57, 128.55, 71.7, 69.8, 69.7, 68.8, 62.7, 62.6, 62.4, 58.8, 58.6, 43.3, 42.1, 39.6, 39.2, 38.9, 38.4, 34.9, 33.1, 33.0, 31.6, 31.4, 29.2, 29.1, 26.5, 26.1, 25.9, 16.6, 16.5, 11.2, 8.0; m/z (FAB) 1216.5982 ([M + H]+ C64H98N3O11P3K requires 1216.6051).
Malonamide ligand 17a. Reaction of amine 3a (1.33 g, 4.35 mmol) with ester 16 (4.67 g 27.9 mmol) gave ligand 17a(1.23 g, 37%). δH(300 MHz; CDCl3) 7.84 (3 H, br s, NH), 3.26–3.48 (30 H, m, CH2OCH2CH2CH2N, C(O)CH2C(O), NCH2C2H4CH3), 3.03, 2.94 (each 4.5 H, 2 × s, NCH3), 1.76 (6 H, quintet, J 6.5, OCH2CH2CH2N), 1.25–1.63 (14 H, m, NCH2C2H4CH3, CH3CH2C), 0.90–0.97 (9 H, m, NCH2C2H4CH3), 0.83 (3 H, t, J 7.5, CCH2CH3); δC(75 MHz; CDCl3) 168.5, 168.4, 166.7, 166.6, 71.7, 69.3, 50.6, 48.0, 43.3, 40.7, 40.2, 37.3, 36.1, 34.0, 30.7, 29.6, 29.4, 23.4, 20.2, 20.1, 14.0, 13.9, 7.9; m/z (FAB) 771.5488 ([M + H]+ C39H75N6O9 requires 771.5596).
Malonamide ligand 17b. Reaction of amine 3b (0.99 g, 2.08 mmol) with ester 16 (3.65 g 21.2 mmol) gave ligand 17b (0.90 g, 46%); δH(300 MHz; CDCl3) 3.23–3.48 (36 H, m, CH2OCH2CH2CH2N, C(O)CH2C(O), NCH2C2H4CH3), 3.05, 3.03, 2.93, 2.92 (together 9 H, 4 × s, NCH3), 1.75–1.86 (6 H, m, OCH2CH2CH2N), 1.25–1.61 (26 H, m, NCH2C2H4CH3, CH3CH2C), 0.89–0.97 (18 H, m, NCH2C2H4CH3), 0.83 (3 H, t, J 7.5, CCH2CH3); m/z (FAB) 939.7297 ([M + H]+ C51H99N6O9 requires 939.7474).
Malonamide ligand 17c. Reaction of amine 3c (1.26 g, 1.95 mmol) with ester 16 (3.50 g 20.32 mmol) gave ligand 17c (1.11 g, 51%); δH(300 MHz; CDCl3) 3.23–3.48 (36 H, m, CH2O, CH2N, C(O)CH2C(O)), 3.04 (5 H, s, NCH3), 2.92–2.93 (4 H, m, NCH3), 1.75–1.86 (6 H, m, OCH2CH2CH2N), 1.47–1.59, 1.20–1.39 (50 H, m, NCH2C2H4CH3, NCH2C6H12CH3, CCH2CH3), 0.81–0.97 (21 H, m, CH2CH3); m/z (FAB) 1145.8746 ([M + K]+ C63H122N6O9K requires 1145.8910).
Malonamide ligand 17d. Reaction of amine 3d (1.415 g, 2.74 mmol) with ester 16 (4.83 g 28.06 mmol) gave ligand 17d (1.50 g, 56%); δH(300 MHz; CDCl3) 4.05–4.19, 3.53–3.67, 3.12–3.48 (33 H, m, CH2O, CH2N, CHN, C(O)CH2C(O)), 3.00–3.02, 2.88–2.90 (together 9 H, m, NCH3), 1.75–1.87 (6 H, m, OCH2CH2CH2N), 1.22–1.56 (26 H, m, (CH3CH2)2CHN, NCH2C2H4CH3, CCH2CH3), 0.78–0.93 (30 H, m, CH2CH3); m/z (FAB) 981.7777 ([M + H]+ C54H105N6O9 requires 981.7943).
Malonamide ligand 18a. Reaction of amine 7a (0.77 g, 3.08 mmol) with ester 16 (6.04 g 32.25 mmol) gave ligand 18a (1.45 g, 66%); δH(300 MHz; CDCl3) 7.96–8.08 (3 H, m, NH), 3.30–3.41 (18 H, m, CH2N, C(O)CH2C(O)), 3.03 (4 H, s, NCH3), 2.95 (5 H, s, NCH3), 2.87–2.98 (6 H, m, PhCH2CH2N), 2.36 (9 H, s, PhCH3), 1.47–1.61, 1.26–1.39 (12 H, m, CONCH2C2H4CH3), 0.91–0.98 (9 H, m, CONCH2C2H4CH3); δC(100 MHz; CDCl3) 168.6, 166.6, 166.5, 133.9, 133.8, 50.7, 48.1, 40.1, 39.7, 38.9, 36.1, 34.1, 30.9, 30.8, 29.4, 20.2, 20.1, 16.2, 14.0; m/z (FAB) 753.4661 ([M + K]+ C39H66N6O6K requires 753.4681).
Malonamide ligand 18b. Reaction of amine 7b (0.49 g, 1.67 mmol) with ester 16 (1.40 g 7.50 mmol) gave ligand 18b (0.84 g, 66%); δH(300 MHz; CDCl3) 8.19–8.29 (3 H, m, NH), 3.31–3.45 (18 H, m, CH2N, C(O)CH2C(O)), 3.06 (4.4 H, s, NCH3), 2.97 (5.6 H, s, NCH3), 2.80–2.90 (6 H, m, PhCH2CH2N), 2.73 (6 H, q, J 6.9, PhCH2CH3), 1.47–1.61, 1.24–1.40 (12 H, m, CONCH2C2H4CH3), 1.16 (9 H, t, J 6.9, PhCH2CH3), 0.92–0.97 (9 H, m, CONCH2C2H4CH3); δC(100 MHz; CDCl3) 168.8, 166.6, 166.5, 140.7, 132.6, 50.7, 48.2, 40.8, 39.9, 39.6, 36.1, 34.1, 30.8, 29.5, 22.7, 20.2, 20.1, 16.1, 13.99, 13.96; m/z (FAB) 795.5157 ([M + K]+ C42H72N6O6K requires 795.5150).
Potentiometric measurements
The procedure for the preparation of the polymeric membranes evaluated for the potentiometric ion response was similar to that described above. The total amount of membrane components was 200 mg and the membranes consisted of 1 wt% of ionophore, 10 mol% of KTFPB (relative to the ionophore) and PVC–o-NPOE (1 ∶ 2 by weight).
Liquid–liquid extractions of americium and europium
2 mL of chloroform were poured into each flask containing the ligand to be tested. After complete dissolution of the latter (quite fast in general), the chloroform solution was separated into 2 batches to perform the liquid–liquid extraction experiments in TCE and n-octanol. Chloroform was dried under a depressurized hood for several days, which allowed the amount of tested ligand to be weighed.500 µL of either TCE or n-octanol were added to dissolve the ligand to be tested and to prepare the organic phases for liquid–liquid extraction experiments. The latter were carried out by contacting 200 µL of the organic samples with 1 and 3 M nitric acid solutions (Vaq = Vorg), spiked with 152Eu(III) and 241Am(III) in 2 mL Eppendorf micro-tubes, thermostated at (25 ± 0.5) °C and shaken for at least 30 min with a vortex IKA device (Vibrax VXR). Tubes were centrifuged and 40 µL of each phase were diluted either with 560 µL of TCE or n-octanol for the organic samples, or with 560 µL of molar nitric acid for the aqueous samples. 550 µL of each sample were taken for radiometric gamma analyses at 59 keV for Am-241 and 121.8 keV for Eu-152, using a Canberra Eurisys pure Ge detector. The acquisition time was long enough to minimize the experimental error: mass balances (Aaqini – (Aaqeq + Aorgeq)/Aaqini, Aaqini, Aaqeq and Aorgeq being, respectively, the activity of the radiotracer in the aqueous phase initially and at equilibrium, and its activity in the organic phase at equilibrium) were always smaller than 10%.
The acidity of the initial and final aqueous solutions was determined by potentiometric titration on 100 µL samples, using a METROHM 751 GPD Titrino device and a 0.1 M NaOH solution.
Acknowledgements
We are grateful for the financial support of the EEC (contract FIKW-CCT-2000-00088) and of the Warsaw University of Technology for the financial support of the work related to the potentiometric part.References and notes
- Managing Radioactive Waste, IAEA factsheet, Vienna, 1998 Search PubMed.
- H. H. Dam, D. N. Reinhoudt and W. Verboom, Chem. Soc. Rev., 2007 10.1039/b603847.
- (a) F. Arnaud-Neu, V. Böhmer, J. F. Dozol, C. Grünter, R. A. Jakobi, D. Kraft, O. Mouauprivez, H. Rouquette, M. J. Schwing-Weill, N. Simon and W. Vogt, J. Chem. Soc., Perkin Trans. 2, 1996, 1175 RSC; (b) L. H. Delmau, N. Simon, M. J. Schwing-Weill, F. Arnaud-Neu, J. F. Dozol, S. Eymard, B. Tournois, V. Böhmer, C. Grünter, C. Musigmann and A. Tunayar, Chem. Commun., 1998, 1627 RSC; (c) S. Barboso, A. Garcia Carrera, S. Matthews, F. Arnaud-Neu, V. Böhmer and M. J. Schwing-Weill, J. Chem. Soc., Perkin Trans. 2, 1999, 264 Search PubMed; (d) A. Arduini, V. Böhmer, L. Delmau, J. F. Desreux, J. F. Dozol, M. A. Garcia Carrera, B. Lambert, C. Musigmann, A. Pochini, A. Shivanyuk and F. Ugozzoli, Chem.–Eur. J., 2000, 6, 2135 CrossRef CAS; (e) C. Schmidt, M. Saadioui, V. Böhmer, V. Host, M.-R. Spirlet, J. F. Desreux, F. Brisach, F. Arnaud-Neu and J. F. Dozol, Org. Biomol. Chem., 2003, 1, 4089 RSC; (f) V. A. Babain, M. Yu. Alyapyashev, M. D. Karavan, V. Böhmer, L. Wang, E. A. Shokova, A. E. Motornaya, I. M. Vatsouro and V. V. Kovalev, Radiochim. Acta, 2005, 93, 749 CrossRef CAS.
- (a) H. Boerrigter, W. Verboom and D. N. Reinhoudt, J. Org. Chem., 1997, 62, 7148 CrossRef CAS; (b) H. Boerrigter, W. Verboom and D. N. Reinhoudt, Liebigs Ann./Recl., 1997, 2247 Search PubMed; (c) H. Boerrigter, W. Verboom, F. De Jong and D. N. Reinhoudt, Radiochim. Acta, 1998, 81, 39 CAS; (d) M. M. Reinoso-Garcia, W. Verboom, D. N. Reinhoudt, F. Brisach, F. Arnaud-Neu and K. Liger, Solvent Extr. Ion Exch., 2005, 23, 425 CrossRef CAS.
- A. Casnati, N. Della Ca, M. Fontanella, F. Sansone, F. Ugozzoli, R. Ungaro, K. Liger and J. F. Dozol, Eur. J. Org. Chem., 2005, 2338 CrossRef CAS.
- (a) E. P. Horwitz, D. G. Kalina, H. Diamond, G. F. Vandegrift and W. W. Schultz, Solvent Extr. Ion Exch., 1985, 3, 75 CrossRef CAS; (b) E. P. Horwitz, K. A. Martin, H. Diamond and L. Kaplan, Solvent Extr. Ion Exch., 1986, 4, 449 CrossRef CAS; (c) E. P. Horwitz, H. Diamond, K. A. Martin and R. Chiarizia, Solvent Extr. Ion Exch., 1987, 5, 419 CrossRef CAS.
- G. J. Lumetta, B. M. Rapko, P. A. Garza, B. P. Hay, R. D. Gilbertson, T. J. R. Weakley and J. E. Hutchison, J. Am. Chem. Soc., 2002, 124, 5644 CrossRef CAS.
- (a) M. W. Peters, E. J. Werner and M. J. Scott, Inorg. Chem., 2002, 41, 1707 CrossRef CAS; (b) K. Matolka, A. Gelis, M. Regalbuto, G. Vandergrift and M. J. Scott, Dalton Trans., 2005, 3719 RSC; (c) V. Rudzevich, D. Schollmeyer, D. Braekers, J. F. Desreux, R. Diss, G. Wipff and V. Böhmer, J. Org. Chem., 2005, 70, 6027 CrossRef CAS; (d) M. M. Reinoso-García, A. Dijkman, W. Verboom, D. N. Reinhoudt, E. Malinowska, D. Wojciechowska, M. Pietrzak and P. Selucky, Eur. J. Org. Chem., 2005, 2131 CrossRef CAS.
- M. M. Reinoso-García, D. Jańczewski, D. N. Reinhoudt, W. Verboom, E. Malinowska, M. Pietrzak, C. Hill, J. Báča, B. Grüner, P. Selucky and C. Grüttner, New J. Chem., 2006, 30, 1480 RSC.
- E. P. Horwitz, D. G. Kalina, L. Kaplan and A. C. Muscatello, Solvent Extr. Ion Exch., 1981, 16, 1127.
- Some recent examples: (a) G. Hennrich and E. V. Anslyn, Chem.–Eur. J., 2002, 8, 2218 CrossRef CAS; (b) A. Vacca, C. Nativi, M. Cacciarini, R. Pergoli and S. Roelens, J. Am. Chem. Soc., 2004, 126, 16456 CrossRef CAS; (c) J. Kim, S.-G. Kim, H. R. Seong and K. H. Ahn, J. Org. Chem., 2005, 70, 7227 CrossRef CAS; (d) J. Kim, B. Raman and K. H. Ahn, J. Org. Chem., 2006, 71, 38 CrossRef; (e) D. R. Turner, M. J. Paterson and J. W. Steed, J. Org. Chem., 2006, 71, 1598 CrossRef CAS.
- (a) L. Nigond, N. Condamines, P. Y. Cordier, J. Livet, C. Madic, C. Cuillerdier, C. Musikas and M. J. Hudson, Sep. Sci. Technol., 1995, 30, 2075 CrossRef CAS; (b) G. Y. S. Chan, M. G. B. Drew, M. J. Hudson, P. B. Iveson, J. O. Liljenzin, M. Skålberg, L. Spjuth and C. Madic, J. Chem. Soc., Dalton Trans., 1997, 649 RSC; (c) G. R. Mahajan, D. R. Prabhu, V. K. Manchanda and L. P. Badheka, Waste Manage. (Amsterdam, Neth.), 1998, 18, 125 Search PubMed; (d) L. Spjuth, J. O. Liljenzin, M. J. Hudson, M. G. B. Drew, P. B. Iveson and C. Madic, Solvent Extr. Ion Exch., 2000, 18, 1 CrossRef CAS; (e) J. N. Mathur, M. S. Murali and K. L. Nash, Solvent Extr. Ion Exch., 2001, 19, 357 CrossRef; (f) Y. Sasaki and S. Tachimori, Solvent Extr. Ion Exch., 2002, 20, 21 CrossRef; (g) S. I. Sinkov, B. M. Rapko, G. J. Lumetta, B. P. Hay, J. E. Hutchison and B. W. Parks, Inorg. Chem., 2004, 43, 8404 CrossRef CAS.
- I. Dayan, J. Libnman, A. Shanzer, C. E. Felder and S. Lifson, J. Am. Chem. Soc., 1991, 113, 3431 CrossRef CAS.
- (a) R. Juday and H. Atkins, J. Am. Chem. Soc., 1955, 77, 4559 CrossRef CAS; (b) M. L. Edwards, N. J. Parkash, D. M. Stemerick, S. P. Sunkara, A. J. Bitonti, G. F. Davis, J. A. Dumont and P. Bey, J. Med. Chem., 1990, 33, 1369 CrossRef CAS; (c) H. Sajiki, T. Ikawa and K. Hirota, Org. Lett., 2004, 6, 4977 CrossRef CAS; (d) R. Nacario, S. Kotakonda, D. M. D. Fouchard, L. M. V. Tillekeratne and R. A. Hudson, Org. Lett., 2005, 7, 471 CrossRef CAS.
- In our case 5% Rh–C14c as a catalyst resulted in a too fast reduction and in addition to the desired secondary amine, also the corresponding primary amine was obtained.
- (a) J. Podlaha, I. Cisarova, D. Alexander, P. Holy, T. Kraus and J. Zavada, Collect. Czech. Chem. Commun., 2000, 65, 1587 CrossRef CAS; (b) B. M. O’Leary, T. Szabo, N. Svenstrup, C. A. Schalley, A. Luezen, M. Schaeer and J. Rebek, J. Am. Chem. Soc., 2001, 123, 11519 CrossRef.
- Due to steric reasons, in these particular cases the secondary amines react less smoothly with the 4-nitrophenyl active ester of CMPO. Therefore the Arbuzov reaction has been used.
- The extraction properties are described with the distribution coefficient, D, defined as the ratio between the metal concentration in the organic and in the aqueous phase at equilibrium. The selectivity toward americium is described by the separation factor, S, defined as the quotient of DAm and DEu.
- S. D. Zaric, Eur. J. Inorg. Chem., 2003, 12, 2197 CrossRef.
- E. Bakker, E. Pretsch and P. Buhlmann, Anal. Chem., 2000, 72, 1127 CrossRef CAS.
- P. Buhlmann, E. Pretsch and E. Bakker, Chem. Rev., 1998, 98, 1593 CrossRef.
- E. Bakker, P. Bühlman and E. Pretsch, Chem. Rev., 1997, 97, 3083 CrossRef CAS.
- P. C. Meier, Anal. Chim. Acta, 1982, 136, 363 CrossRef CAS.
- Y. Mi and E. Bakker, Anal. Chem., 1999, 71, 5279 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2007 |