Stabilization of terpyridine covered gold nanoparticles by metal ions complexation
Marco
Montalti
*a,
Luca
Prodi
a,
Nelsi
Zaccheroni
a,
Matteo
Beltrame
a,
Tamara
Morotti
b and
Silvio
Quici
b
aChemistry Department “G. Ciamician”, Latemar Unit, University of Bologna, Via Selmi 2, 40126, Bologna, Italy. E-mail: marco.montalti2@unibo.it; Fax: +39-051 2099456
bCNR—Istituto di Scienze e Tecnologie Molecolari, Via C. Golgi 19, 20133, Milano, Italy
First published on 16th October 2006
Abstract
The introduction of 1, a thiol presenting a terpyridine ligand, on the surface of weakly stabilized nanoparticles leads, at high concentration of this capping agent, to the aggregation of the gold clusters. This behaviour is due to the weak, but not negligible interaction of the terpyridine moiety with the metal surface, an interaction that becomes relevant at high concentrations of 1 on the surface, when several units can cooperate in interlinking the nanoparticles. To avoid this effect, we have investigated the complexation of zinc by the terpyridine derivative bound to gold nanoclusters, in excess of metal ion and in the presence of coordinating anions, as a powerful tool for the stabilization of gold nanoparticles. In this context, anions, such as bromide, showed to play an important role in this processes; the use of anions containing desired functions can than be a very versatile strategy to obtain new enriched nanoparticles.
Introduction
The research on gold nanoparticles is, at present, one of the most fertile topics in chemistry and in life and material sciences. This is mostly due to the versatility of these systems; the properties of gold nanoparticles, in fact, can be tuned in a huge range thanks to the combination of their particular and unusual optical and electronic characteristics1 with the functionalities that is possible to introduce on their surface.2 All these possibilities open up new intriguing perspectives in the design of functional nanosystems with promising applications in the fields of diagnostics, electronics and optics.3 | ||
| Scheme 1 | ||
The stability of gold nanoparticles is typically ensured by capping agents that strongly bind to their surfaces via a gold–thiol bond,2,4 thus preventing their irreversible aggregation.5 Other capping materials like oxoanions, amine or thiocyanate have also been used for nanoparticles functionalization, solubilization, and stabilization.2,6 Also polypyridines, in particular terpyridine, have showed to be suitable units for anchoring fullerene to gold nanoparticles.7 It is important to stress that the variety of possible binding units makes the chemistry of gold nanoparticles extremely versatile, provided that all possible processes could be carefully controlled. Molecules conceived for gold nanoparticles functionalization, in fact, usually contain an active part (a fluorophore, a receptor or an electro-active moiety) tailed with a thiolic group. This last part is of course the one designed for the binding to the gold nucleus, while a direct interaction of the active unit to the gold surface is usually undesirable since it may cause the loss of its specific function, cross-linking, or even irreversible aggregation of the nanoparticles. Although cross linking has been recently exploited as an ingenious strategy for sensing or for the preparation of nanostrucured materials,2,3,8 it is often necessary to have a stable, not aggregated, dispersion of nanoparticles. In this paper, we present a detailed study about the use of compound 1, which presents both a thiol and a terpyridine unit, as a capping agent for gold nanoclusters. The organization of 1 on gold flat macroscopic surfaces has been already investigated;9 however, its behaviour on nanoparticles’ surface is of special interest. In particular, we show in which conditions 1 causes aggregation of the nanoparticles and how it is possible to avoid this process. It is to note that, as we revised the present article, Zhang et al. presented, on the contrary, the possibility to form large 3D and highly dispersed spherical assemblies among nanoparticles using a very similar terpyridine containing thiol as capping agent.10
Our approach to the problem is based on the high sensitivity offered by fluorescence spectroscopy,11 and it takes advantage from the results described in a previous work12 where we showed how it is possible to control the gradual coverage of preformed TOAB (tetraoctylammounium bromide) stabilized nanoparticles in solution adding defined amounts of a thiol functionalized with the pyrene chromophore. The assembly of the capping layer could be followed in real time even in very dilute solutions via fluorescence spectroscopy, in fact, it is usually possible to distinguish between the bound and unbound fluorophores even at the subnanomolar level. In this way, because of the high dilution, also species that typically cause irreversible aggregation processes can still be characterized. Hence it becomes possible to obtain quantitative information about stability in solution that could not be gained otherwise. We would like to stress that the purpose of this work is not merely to prepare nanoparticles bearing the 1 molecules on the surfaces which could be done via a thiol exchange procedure,2,8c,13 but to understand and control the different interactions of the thiol group and the terpyridine moiety with the gold nanoparticles and to control in this way the aggregation of nanoparticles.
Experimental
Synthesis of Gold nanoparticles
All the reagents and solvents were purchased by Sigma-Aldrich and used without further purification. For the preparation of the gold clusters we slightly modified the original procedure proposed by Brust et al.4 Briefly 1 ml of a water solution of HAuCl4 (1.3 × 10−2 M) was stirred together with 1 ml of toluene solution of TOAB (4.0 × 10−2 M). After 1 h, the complete transfer of the [AuCl4]− anion to the organic phase was confirmed by colour changes of the two phases: the initially yellow water solution became completely colourless, while toluene turned to deep red. After separation of the two phases, 1 ml of NaBH4 water solution (0.1 M) was stirred for 3 h together with the toluene solution containing the [AuCl4]− anion. After gold reduction a deeply purple solution of metal nanoparticles resulted and their average diameter was measured by TEM. Compound 1 was available from previous work.9Spectroscopy
Absorption spectra were recorded on a Perkin-Elmer lambda 40 spectrophotometer.Fluorescence spectra (450 W Xe lamp), excited state lifetimes (pulsed laser at 406 nm), steady state and time resolved anisotropy measurements were obtained with a modular UV-vis-NIR spectrofluorimeter Edinburgh, equipped with single-photon counting apparatus. Correction of fluorescence spectra for taking into account the absorption of gold nanoparticles at the excitation and emission wavelengths has been performed as previously reported.14
Results and discussion
Nanoparticle experiments
The addition of increasing amounts of 1 to a 6 × 10−9 M acetonitrile solution of TOAB stabilized gold nanoparticles leads to the appearance, in the absorption spectrum, of the band at 287 nm typical of the polypyridinic compound (Fig. 1). | ||
| Fig. 1 Changes in the absorption spectrum of a 6 × 10−9 M acetonitrile gold nanoparticles solution upon addition of 1 (between 0 and 3 × 10−6 M). See Scheme 2 for the meaning of I and II. | ||
The presence of 1, on the other hand, causes only minor changes on the absorbance of the plasmon resonance band of the metal nanoparticles provided that its concentration is sufficiently small (i.e., less than 2 × 10−6 M). A big red shift and intensification of the plasmon resonance band is instead observed in the presence of a higher concentration of 1. Finally, after reaching a critical concentration of the capping agent (3.5 × 10−6 M), the trend observed for the absorption in the visible region is reversed and the intensity of the band decreases. These changes are clearly visible in Fig. 1 and 2 and can be easily followed by reporting the value of absorption at 600 nm as a function of the concentration of 1 as in Fig. 3, where three distinct regions with different behaviours can be clearly spotted. In the low concentration region (below 2 × 10−6 M, range I in Fig. 3) the perturbation of the plasmon resonance band due to the addition of 1 is small, indicating a low degree of aggregation of the colloids. In the medium concentration region (between 2 × 10−6 M and 3.0 × 10−6 M, range II in Fig. 3) a strong aggregation causes a large increase of the absorbance followed, at higher concentration (over 3.0 × 10−6 M, range III in Fig. 3) by the formation of big aggregates that after several minutes precipitate from the solution as a black powder.
 | ||
| Fig. 2 Changes in the absorption spectrum of a 6 × 10−9 M acetonitrile gold nanoparticles solution upon addition of 1 (between 3 × 10−6 M and 5 × 10−6 M). See Scheme 2 for the meaning of II and III. | ||
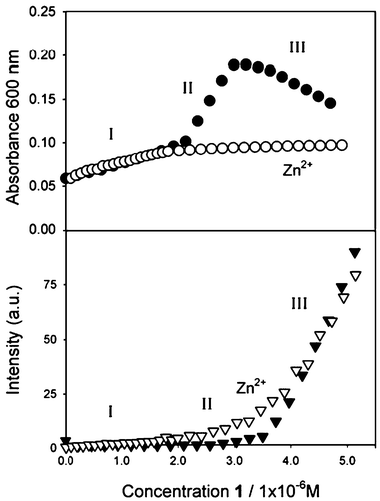 | ||
| Fig. 3 Changes in the absorption and fluorescence intensity of a 6 × 10−9 M acetonitrile solution of gold nanoparticles upon addition of 1. The concentration range I, II and III correspond to those described in Scheme 2. Top: full circle and empty circles correspond to the absence and the presence of zinc (2 × 10−4 M), respectively. Bottom: fluorescence intensity (λexc = 310 nm, λem = 405 nm) in the absence of zinc is represented by the full triangles; fluorescence intensity (λexc = 310 nm, λem = 500 nm) in the presence of zinc (2 × 10−4 M) is represented by the empty triangles. | ||
The binding of the thiol 1 to the gold surface can also be followed by detecting its fluorescence. The fluorescence of the terpyridine is, in fact, dramatically quenched when bound to the gold surface and only a very weak band is detected for concentrations under the surface saturation level (below 3.5 × 10−6 M) while the fluorescence observed at higher concentrations is mostly due to the presence of unbound 1 molecules. Fig. 4 clearly shows how the fluorescence intensity at 405 nm has a large increase only after the complete coverage of the nanoparticles surfaces, corresponding to the increase of the concentration of the free fluorophores. Taking into account the concentration of 1 at saturation (3.5 × 10−6 M) and the mean radius of nanoparticles obtained from TEM experiments, it is possible to calculate that the average surface area occupied by each chromophore is 25 Å2. It is to note that this value is comparable with those reported for similar thiolic derivatives.13 The correlation between the absorption and fluorescence signals can be examined in Fig. 3. As already observed, the fluorescence data indicates that full coverage of the available nanoparticles surfaces takes place for a 3.5 × 10−6 M concentration of 1. On the other hand, the changes in absorbance show that aggregation of the nanoparticles starts at a concentration around 2 × 10−6 M and that it becomes so strong to cause a gradual disappearing of the plasmon resonance band when a concentration 3 × 10−6 M is reached. It is interesting to note that the two signals, namely the fluorescence intensity and the absorbance, allow us to follow the binding and aggregation processes from the point of view of the fluorophore and of the nanoparticles, respectively, confirming the richness of information obtainable via optical investigation. In order to simplify the understanding of the behaviour of the system discussed in this section, the species present in solutions in the concentration ranges I, II and III of Fig. 3 are represented in Scheme 2.
 | ||
| Scheme 2 Schematic representation of the interaction of 1 with gold nanoparticles: I corresponds to low concentration of 1 (roughly less than 50% of surface coverage). II is the medium concentration regime (between 50 and 100% of surface coverage). III corresponds to an excess of 1. The pictures above the roman numbers describe the aggregation observed in the absence of zinc. The pictures below the numbers are relative to the presence of zinc. In the case of II the saturated (100% coverage) nanoparticles are represented. In both cases (with and without zinc) strong quenching of the fluorescence of 1 is observed in cases I and II and strong fluorescence coming from unbound molecules (or complexes) in the of case III. | ||
 | ||
| Fig. 4 Fluorescence spectra (λexc = 310 nm) of a 6 × 10−9 M acetonitrile solution of gold nanoparticles upon addition of 1 (concentration between 0 and 5 × 10−6 M). See Scheme 2 for the meaning of I and III. | ||
Summarizing, the attempt to cover the surface of the nanoparticles with 1 causes their irreversible aggregation. This is not surprising when the possible nature of 1 as a crosslinker agent is considered and is in agreement with the failure of our attempts to synthesize stable gold nanoparticles by reduction of gold in the presence of 1 in the conditions proposed by Brust and coworkers.4 What is surprising, on the contrary, is that aggregation occurs only after a threshold concentration is exceeded. This means that a stable solution of gold nanoparticles having 1 molecules bound to the surface can be easily prepared if the loading of organics is kept under the aggregation limit. A possible explanation of this peculiar behaviour is that terpyridine moieties ‘diluted’ on the surface are ineffective and that a critical local density of terpyridine units has to be reached so that they can cooperate in cross linking the nanoparticles.
Complexation experiments
The well known properties of terpyridine as metal chelators13 suggested a direct strategy for controlling the process: in particular we though that ‘locking’ the coordinative site with a suitable metal ion in appropriate experimental conditions aggregation of the metal nanoparticles could be completely avoided. In particular, it was shown by the group of Rotello and, very recently, by the one of Li that the use of Fe2+, Zn2+, Co2+, Ag+, and Cu+ can lead to large assemblies of nanoparticles derivatized with terpyridine ligands. We choose to work with Zn2+ ions, whose coordination with terpyridine derivatives has been widely investigated,15 because they are known to adopt also coordination numbers lower than 6.16 For this reason, we started to study the coordination ability of 1 towards this metal ions in different experimental conditions.In particular, we started following the spectrophotometric and fluorimetric titrations of 1 with zinc triflate in order to look at the stoichiometry and the photophysical properties of its zinc complexes. Compound 1 presents in acetonitrile solution a strong absorption band at 287 nm with a molar absorbivity ε = 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 (Fig. 5a). The fluorescence spectrum (Fig. 5c) shows a band with maximum at 405 nm, and excited state lifetime of 1.5 ns. Upon addition of Zn2+ the absorption and fluorescence bands of 1 shift to longer wavelengths (Fig. 6 and 7) indicating the formation of both [1·Zn]2+ and [12·Zn]2+ complexes, according to eqn (1) and (2).
000 M−1 cm−1 (Fig. 5a). The fluorescence spectrum (Fig. 5c) shows a band with maximum at 405 nm, and excited state lifetime of 1.5 ns. Upon addition of Zn2+ the absorption and fluorescence bands of 1 shift to longer wavelengths (Fig. 6 and 7) indicating the formation of both [1·Zn]2+ and [12·Zn]2+ complexes, according to eqn (1) and (2).
| 1 + Zn2+ ↔ [1·Zn]2+ | (1) |
| [1·Zn]2+ + 1 ↔ [12·Zn]2+ | (2) |
![Curves a and b are the absorption spectra of 1 and of its zinc complex [1·Zn]2+, respectively. Lines c and d are the fluorescence spectra (λexc = 310 nm) of 1 and [1·Zn]2+, respectively. Fluorescence spectrum e was recorded for a 1 × 10−6 M acetonitrile solution of 1 in the presence of TOAB (6 × 10−4 M) and zinc triflate (2 × 10−4 M).](/image/article/2007/NJ/b600339g/b600339g-f5.gif) | ||
| Fig. 5 Curves a and b are the absorption spectra of 1 and of its zinc complex [1·Zn]2+, respectively. Lines c and d are the fluorescence spectra (λexc = 310 nm) of 1 and [1·Zn]2+, respectively. Fluorescence spectrum e was recorded for a 1 × 10−6 M acetonitrile solution of 1 in the presence of TOAB (6 × 10−4 M) and zinc triflate (2 × 10−4 M). | ||
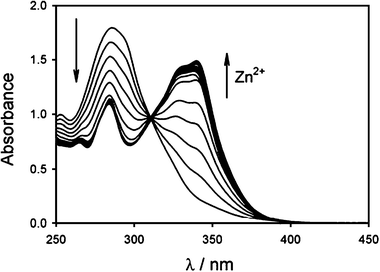 | ||
| Fig. 6 Changes in the absorption spectrum of a 5 × 10−5 M acetonitrile solution of 1 during titration with zinc triflate. The last spectrum corresponds to the addition of 1.5 equivalents of zinc. | ||
 | ||
| Fig. 7 Changes in the fluorescence spectrum (λexc = 310 nm) of a 5 × 10−5 M acetonitrile solution of 1 during titration with zinc triflate. The last spectrum corresponds to the addition of 1.5 equivalents of zinc. | ||
Stabilization of the nanoparticles by zinc complexation
Carrying out the experiment with nanoparticles discussed in the previous paragraph in the presence of zinc triflate in excess (2 × 10−4 M), a negligible aggregation of the nanoparticles is observed. As it can be seen from Fig. 8, in fact, the addition of 1 induces only a small change of the plasmon resonance band of the nanoparticles, an expected change considering the modification of the surface properties caused by the thiol binding. The stabilizing effect of zinc ions is even more clear in Fig. 4 where the changes in the absorption at 600 nm are compared with those observed in the absence of the zinc salt: no significant variations takes place in the ‘critical’ concentration regions II and III. This result suggests that in the presence of an excess of zinc the terpyridine function is already involved in metal complexation, a conclusion that can be confirmed by combining the absorption and fluorescence data. Fig. 4 and 9 in fact clearly show that the fluorescence of 1 is strongly quenched by the gold nanoparticles: such an efficient quenching is not compatible in the experimental conditions with a diffusional process and can occur only upon binding of the fluorescent molecules to the gold. The complexation of zinc ions, on the other hand, is revealed primarily by the presence of the absorption band of the complex at 340 nm but also by the spectral position of the residual fluorescence band (Fig. 9). The position of the fluorescence maximum at 465 nm, typical of the [1·ZnBr2] complex (Fig. 5), suggests also that the presence of bromide anions, introduced during the colloids preparation, plays a fundamental role in the stabilization effect, inducing an electric neutrality to the complex and helping to avoid cross linking expected by the formation of [12·Zn]2+ complexes. From the TEM and titration data it has been possible to calculate again an average surface area occupied by each complex of 25 Å2.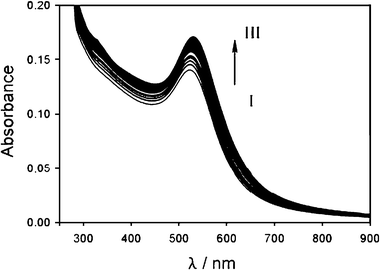 | ||
| Fig. 8 Changes in the absorption spectrum of a 6 × 10−9 M acetonitrile solution of gold nanoparticles upon addition of 1 (concentration between 0 and 3 × 10−6 M) in the presence of zinc triflate 2 × 10−4 M. See Scheme 2 for the meaning of I and III. | ||
 | ||
| Fig. 9 Fluorescence spectra (λexc = 310 nm) of a 6 × 10−9 M acetonitrile solution of gold nanoparticles upon addition of 1 (concentration between 0 and 5 × 10−6 M) in the presence of zinc triflate 2 × 10−4 M. See Scheme 2 for the meaning of I and III. | ||
It is interesting to note that, despite of their important stabilizing effect, zinc ions do not alter the affinity of 1 for gold nanoparticles. The fluorescence intensity curves of Fig. 4 that allow to follow the process of binding in the presence or in the absence of zinc are in fact very similar to each other. Even the average surface area occupied by the fluorophore, calculated by the surface saturation point, is the same whether the terpyridine is complexed or not.
The data that we present here show a very interesting feature of this system: while ligand 1 alone is not able to stabilize the colloids, leading to their irreversible aggregation when a critical surface concentration is reached, its zinc complex in suitable conditions prevents this phenomenon. This suggests that the complexation process can be exploited also as a possible mean for nanoparticle stabilization.
Moreover, the possibility to change the coordinated anions, using, for example, functionalized carboxylates,19 can afford an additional and versatile tool to prepare nanostructures having desired properties.
Together with the stability effect, it is also interesting to note the photophysical behaviour of this system. This is in fact a further example of fluorescence quenching near the surface of gold nanoparticles. The investigation of the mechanism of this process is beyond the purpose of this paper, but the fact that also the metal complex with zinc of 1 is quenched by the interaction with the gold nanoparticles suggests a process quite independent from the electrochemical properties of the fluorophore. Quenching via energy transfer seems than to be more likely than electron transfer, especially if one considers that, in the case of nanoparticles, this process is much less sensitive to the energy of the excited state of the donor with respect to molecular quenchers.20
We would like also to stress here that combined UV-vis and fluorescence spectroscopy allow a detailed monitoring of the ‘state’ of each component of the system. The degree of aggregation is revealed by the changes in the characteristic plasmon resonance band of the colloids1,2 while the fraction of 1 molecules effectively bound to the gold can be measured by fluorescence spectroscopy since the interaction with the nanoparticles dramatically quenches its fluorescence.
Conclusions
The results obtained allowed us to conclude that a low concentration of 1 on the gold nanoparticles does not alter their stability while, when a critical concentration value is reached, aggregation takes place. This indicates that, while, as expected, the binding of the thiol to the gold is strong and almost quantitative even at very low concentrations, the interaction with the terpyridine moiety is much weaker and it becomes relevant only at higher concentrations when several units can cooperate in interlinking the nanoparticles. We demonstrated that complexation of zinc by terpyridine on the surface of gold colloids is not only possible, but that it also strongly contributes to stabilize, in suitable experimanetal conditions, the nanoparticles in solutions. Anions, such as bromide, showed to play an important role in this stabilization processes. The growth of metal complexes on colloids surface can be hence considered a new strategy for modifying the surface of nanoparticles. A suitable choice of the anions could also allow to design more versatile structures via the binding of negatively charged substrates possessing desired chemical functions.Acknowledgements
This work has been supported by MIUR through FIRB 2003–2004 LATEMAR (www.latemar.polito.it) and FIRB RBNE019H9K (Molecular Manipulation for Nanometric Machines) and by CNR.References
- See for example: (a) D. Lee, R. L. Donkers, G. Wang, A. S. Harper and R. W. Murray, J. Am. Chem. Soc., 2004, 126, 6193–6199 CrossRef CAS; (b) R. Guo and R. W. Murray, J. Am. Chem. Soc., 2005, 127, 12140–12143 CrossRef CAS; (c) G. Wang, T. Huang, R. W. Murray, L. Menard and R. G. Nuzzo, J. Am. Chem. Soc., 2005, 127, 812–813 CrossRef CAS.
- (a) A. C. Templeton, W. P. Welfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS; (b) P. V. Kamat, J. Phys. Chem. B, 2002, 106, 7729–7744 CrossRef CAS; (c) K. G. Thomas and P. V. Kamat, Acc. Chem. Res., 2003, 36, 888–898 CrossRef CAS; (d) R. Shenhar and V. M. Rotello, Acc. Chem. Res., 2003, 36, 549–561 CrossRef CAS; (e) M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS; (f) U. Drechsler, B. Erdogan and V. M. Rotello, Chem.–Eur. J., 2004, 10, 5570–5579 CrossRef CAS.
- For some recent example see: (a) C. S. Thaxton, H. D. Hill, D. G. Georganopoulou, S. I. Stoeva and C. A. Mirkin, Anal. Chem., 2005, 77, 8174–8178 CrossRef CAS; (b) S. I. Stoeva, F. Huo, J.-S. Lee and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 15362–15363 CrossRef CAS; (c) L. Wang, R. Yan, Z. Huo, L. Wang, J. Zeng, J. Bao, X. Wang, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 6054–6057 CrossRef CAS; (d) F. Seker, P. R. L. Malenfant, M. Larsen, A. Alizadeh, K. Conway, A. M. Kulkarni, G. Goddard and R. Garaas, Adv. Mater., 2005, 17, 1941–1945 CrossRef CAS.
- M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, J. Chem. Soc., Chem. Commun., 1995, 1655–1656 RSC.
- See for example: K. Aslan and V. H. Pérez-Luna, Langmuir, 2002, 18, 6059–6065 Search PubMed.
- (a) S. L. Cumberland and G. F. Strouse, Langmuir, 2002, 18, 269–276 CrossRef CAS; (b) S. R. Isaacs, E. C. Cutler, J.-S. Park, T. R. Lee, T. R. and Y.-S. Shon, Langmuir, 2005, 21, 5689–5692 CrossRef CAS; (c) K. G. Thomas and P. V. Kamat, J. Am. Chem. Soc., 2000, 122, 2655–2656 CrossRef CAS; (d) J. Turkevich, P. C. Stevenson and J. Hiller, Discuss. Faraday Soc., 1951, 11, 55 RSC; (e) K. G. Thomas, J. Zajicek and P. V. Kamat, Langmuir, 2002, 18, 3722–3727 CrossRef CAS.
- H. Fang, C. Du, S. Qu, Y. Li, Y. Song, H. Li, H. Liu and D. Zhu, Chem. Phys. Lett., 2002, 364, 290–296 CrossRef CAS.
- See for example: (a) Y. Kim, R. C. Johnson and J. T. Hupp, Nano Lett., 2001, 1, 165–167 CrossRef CAS; (b) S. Lin, M. Li, E. Dujardin, C. Girard and S. Mann, Adv. Mater., 2005, 17, 2553–2559 CrossRef CAS; (c) B. N. Tyler, L. F. Benjamin and V. M. Rotello, Nano Lett., 2002, 2, 1345–1348 CrossRef CAS; (d) X. Zhang, D. Li and X.-P. Zhou, New J. Chem., 2006, 30, 706–715 RSC; (e) R. Baron, C.-H. Huang, D. M. Bassani, A. Onopriyenko, M. Zayats and I. Willner, Angew. Chem., Int. Ed., 2005, 44, 4010–4015 CrossRef CAS.
- A. Auditore, N. Tuccitto, G. Marzanni, S. Quici, F. Puntoriero, S. Campagna and A. J. Licciardello, Chem. Commun., 2003, 2494–2495 RSC.
- X. Zhang, D. Li and X.-P. Zhou, New J. Chem., 2006, 30, 706–711 RSC.
- L. Prodi, New J. Chem., 2005, 29, 20–31 RSC.
- M. Montalti, L. Prodi, N. Zaccheroni and G. Battistini, Langmuir, 2004, 20, 7884–7886 CrossRef CAS.
- (a) R. Guo, Y. Song, G. Wang and R. W. Murray, J. Am. Chem. Soc., 2005, 127, 2752–2757 CrossRef CAS; (b) R. L. Donkers, Y. Song and R. W. Murray, Langmuir, 2004, 20, 4703 CrossRef CAS; (c) G. H. Woehrle and J. E. Hutchison, Inorg. Chem., 2005, 44, 6149 CrossRef CAS; (d) G. H. Woehrle, L. O. Brown and J. E. Hutchison, J. Am. Chem. Soc., 2005, 127, 2172–2183 CrossRef CAS; (e) M. Montalti, L. Prodi, N. Zaccheroni, R. Baxter, G. Teobaldi and F. Zerbetto, Langmuir, 2003, 19, 5172–5174 CrossRef CAS.
- (a) A. Credi and L. Prodi, Spectrochim. Acta, Part A, 1998, 54, 159–170 CrossRef; (b) M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, 3rd edn., CRC Taylor and Francis, Boca Raton, 2006 Search PubMed.
- See for example: (a) F. Loiseau, C. Di Pietro, S. Serroni, S. Campagna, A. Licciardello, A. Manfredi, G. Pozzi and S. Quici, Inorg. Chem., 2001, 40, 6901–6909 CrossRef CAS; (b) M. Schmittel, V. Kalsani, R. S. K. Kishore, H. Cölfen and J. W. Bats, J. Am. Chem. Soc., 2005, 127, 11544–11545 CrossRef CAS; (c) C. Bazzicalupi, A. Bencini, A. Bianchi, A. Danesi, C. Giorgi, C. Lodeiro, F. Pina, S. Santarelli and B. Valtancoli, Chem. Commun., 2005, 2630–2632 RSC; (d) M. Barboiu, L. Prodi, M. Montalti, N. Zaccheroni, N. Kyritsakas and J.-M. Lehn, Chem.–Eur. J., 2004, 10, 2953–2959 CrossRef CAS.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, 2nd edn, Elsevier, Amsterdam, 1997 Search PubMed.
- The titration data were analized with SPECFIT by R. A. Binstead, Spectrum Software Associates, Chapell Hill, NC Search PubMed.
- F. Loiseau, C. Di Pietro, M. Cavazzini, G. Marzanni and S. Quici, J. Mater. Chem., 2005, 15, 2762–2771 RSC.
- (a) L. Prodi, R. Ballardini, M. T. Gandolfi and R. Roversi, J. Photochem. Photobiol., A, 2000, 136, 49–52 CrossRef CAS; (b) A. Juris and L. Prodi, New J. Chem., 2001, 25, 1132–1135 RSC.
- (a) J. Gersten and A. Nitzan, J. Chem. Phys., 1981, 75, 1139–1152 CrossRef CAS; (b) C. Fan, S. Wang, J. W. Hong, G. C. Bazan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6297–6301 CrossRef CAS; (c) E. Dulkeith, A. C. Morteani, T. Niedereichholz, T. A. Klar, J. Feldmann, S. Levi, F. C. J. M. van Veggel, D. N. Reinhoudt, M. Möller and D. I. Gittins, Phys. Rev. Lett., 2002, 89, 203002 CrossRef CAS; (d) E. Dulkeith, M. Ringler, T. A. Klar, J. Feldmann, A. Muñoz Javier and W. J. Parak, Nano Lett., 2005, 5, 585–589 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2007 |