A novel electrochemically synthesized biodegradable thin film of polypyrrole–polyethyleneglycol–polylactic acid nanoparticles
Galit
Shustak
ab,
Mariusz
Gadzinowski
c,
Stanislaw
Slomkowski
c,
Abraham J.
Domb
b and
Daniel
Mandler
*a
aDepartment of Inorganic and Analytical Chemistry, The Hebrew University of Jerusalem, Jerusalem 91904, Israel. E-mail: mandler@vms.huji.ac.il; Fax: +972-2-6585319; Tel: +972-2-6585831
bDepartment of Medicinal Chemistry and Natural Products, School of Pharmacy-Faculty of Medicine, The Adolph Weinberger Building, P.O. Box 12065, The Hebrew University of Jerusalem, Jerusalem 91120, Israel. E-mail: adomb@md.huji.ac.il; Fax: +972-2-6757629; Tel: +972-2-675-7573
cCentre of Molecular and Macromolecular Studies, Laboratory of Biomedical Polymers, Sienkiewicza 112, 90-363 Lodz, Poland
First published on 17th October 2006
Abstract
Nanoparticles having reactive pyrrole residues were prepared from poly(1-ethoxyethylglycidyl ether)-block-poly(L,L-lactide) block copolymer. The nanoparticles were electropolymerized in aqueous media through the oxidation of the pyrrole residue and in the presence of pyrrole to form a nanocomposite thin film. The novel synthesis of these pyrrole-functionalized nanoparticles is described and the electrochemical deposition of the corresponding coating is characterized using electrochemistry, SEM and EDX.
Introduction
Electrically conducting polymers have attracted much interest in the last twenty years because they combine the physical and chemical properties of organic polymers with the electric characteristics of metals. Among the known conducting polymers, polypyrrole has probably been the most investigated and applied, because of its simple method of preparation, high conductivity and durability.1,2 Polypyrrole has been frequently utilized in biochemistry and medicine as a matrix for assembling biosensors.3–10 Moreover, the reversible doping–undoping mechanism of polypyrrole has been the driving force for the delivery of charged biological substances to and from the polymer matrix.11–15 For example, Miller and Zhou showed that a copolymer film was doped with dopamine upon its reduction.12 The entrapped dopamine was released by stepping the polymer to a positive potential.The chemical and physical properties of conducting polymers, such as redox activity, morphology and conductivity, can be tuned by incorporating nanoparticles into the polymeric matrix.5,16–34 For example, the conductivity of polyaniline and polypyrrole was markedly improved upon embedding gold and silver nanoparticles.17,33 Lee and Liu reported that pyrrole, which was catalytically electrooxidized upon incorporating silver nanoparticles into the polypyrrole matrix, showed superior conductivity.16 Nanoparticle-doped conducting polymers can also be used as electrochemical sensors5,34 as has been demonstrated by Xian and coworkers, who described a glucose biosensor based on gold nanoparticles embedded in polyaniline nanofibers.5
To date, several different approaches have been employed for the preparation of metal or inorganic nanoparticle-conducting polymer nanocomposites, such as chemical polymerization, physical mixing,35 layer-by-layer assembly17 and sol–gel deposition.36 Only a few studies have focused on the incorporation of organic nanoparticles into conducting polymers.37–39 Shi and coworkers synthesized a conductive polypyrrole nanoparticle–poly(D,L-lactide) composite on which the growth of fibroblasts was regulated by direct electron current.37
In spite of the fact that organic nanoparticles can be formulated from a wide variety of synthetic and natural polymers, biodegradable polymers are especially appealing due to their biodegradability and biocompatibility with cells and tissues.40 Nanoparticles of biodegradable polymers, such as polylactic acid (PLA) and polyethyleneglycol (PEG)-modified PLA, have recently been used for sustained and localized administration of different therapeutic agents.41–44
In this study we report the formation and characterization of a thin polypyrrole matrix to which PLA nanoparticles are covalently attached via electropolymerization in a single step. Firstly, PLA nanoparticles having reactive pyrrole residues were synthesized from poly(1-ethoxyethylglycidyl ether)-block-poly(L,L-lactide) block copolymer. In a subsequent step, the modified PLA nanoparticles were electropolymerized on 316L stainless steel and evaporated gold electrodes in the presence of pyrrole. Such a thin polypyrrole film containing biodegradable nanoparticles can be used for coating implantable medical devices usually made of 316L stainless steel. The advantages of the approach described here include highly biocompatible organic material, large drug loading (compared to polymer swelling for example) and good control of drug release. These features can be tailored by controlling the size, shape, and functions of the nanoparticle building blocks.
Materials and methods
Materials
Pyrrole (98%) was purchased from Sigma-Aldrich and was freshly purified by alumina column chromatography before use. Alumina for column chromatography was purchased from ICN Biomedicals, Germany (activity grade I). NaF (99%) was obtained from BDH Chemicals and used as received. Deionized water (Barnstead Easypure UV system) was used for preparing the different solutions. Acetonitrile (ACN, >99.8%) was obtained from J. T. Baker while acetone and dichloromethane HPLC grade were from BioLab (Jerusalem, Israel).L,L-Lactide (Boehringer, Germany) was purified by crystallization from 2-propanol and subsequent sublimation. Purified monomer was protected from contact with air by being stored in an evacuated ampoule. 1-Ethoxyethylglycidyl ether was synthesized by reaction of glycidol (Aldrich) and ethyl vinyl ether (Fluka) according to the known procedure.45 The initiator potassium tert-butoxide (Aldrich) was used as received. Tetrahydrofuran (THF, POCh, Poland) was distilled and dried over Na wires. Thereafter, these solvents were dried over a Na–K alloy in ampoules equipped with Teflon® stopcocks and stored under vacuum. The required amounts of THF were distilled using a vacuum line to polymerization vessels. 1,4-Dioxane (POCh, Poland) was purified by distillation. The fraction boiling in a range of 100–102 °C was collected. Methylene chloride (POCh, Poland) was purified according to the following procedure. First, solvent was stirred with concentrated sulfuric acid (10% v/v with respect to the solvent). Thereafter, methylene chloride was isolated using a separating funnel and washed with several portions of distilled water. Then it was preliminarily dried with CaCl2, distilled (the fraction boiling at 40 °C was collected) and dried in an evacuated ampoule over P2O5. The necessary amount of methylene chloride was distilled prior to use.
Syntheses
Signals in the 1H NMR spectrum of I were assigned as follows: 1.0–1.3 (overlapping s and d, (CH3)3C– end-group from initiator and CH3CH<, polyEEGly); 1.55 (t, –CO(CHCH3)O–, PLLA); 3.20–3.85 (m, –OCH2–, –OCH<; polyEEGly); 4.64 (q, –OCH(CH3)O–, polyEEGly); 5.05–5.25 (m,–COCH(CH3)O–, PLLA).
The hydroxyl groups were deprotected by dissolving I in 1,4-dioxane : water 8 : 2 (v/v) mixture (ca. 150 mL) to which 20 mL of concentrated formic acid was added. The mixture was stirred for 4 days. Then it was frozen and lyophilized to give polyglycidol–block-poly(L,L-lactide) (II).
Signals in the 1H NMR spectrum of III were assigned as follows: 1.15 (s, (CH3)3C– end-group from initiator; 1.56 (d, overlapping doublets of −C(O)CH(CH3)O– groups along the chain and end groups of PLLA blocks), 2.65–2.90 (t, –CH2CH2C(O)–, labeled polyGly); 3.30–3.65 (m, –OCH2–, –OCH<, polyGly labeled and not labeled, >CHCH2OH, polyGly); 3.9–4.4 (m, >CHCH2O– and >NCH2CH2–, labeled polyGly); 5.25 (q, –C(O)CH(CH3)O–, PLLA); 6.05 (d, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHN–, pyrrole); 7.23 (d, –CHCHN–, pyrrole). Integration of signals in the spectrum was in agreement with the number average degree of polymerization of the PLLA block (2m + 1) being equal to 42 (Mn = 3000), of the labeled polyGly block (k) equal to 24 (Mn = 4700) and of the unlabeled polyGly segment (n + k) being equal to two. Thus, the efficiency of labeling was ca. 92%.
CHN–, pyrrole); 7.23 (d, –CHCHN–, pyrrole). Integration of signals in the spectrum was in agreement with the number average degree of polymerization of the PLLA block (2m + 1) being equal to 42 (Mn = 3000), of the labeled polyGly block (k) equal to 24 (Mn = 4700) and of the unlabeled polyGly segment (n + k) being equal to two. Thus, the efficiency of labeling was ca. 92%.
Characterization
The surface morphology of the modified electrodes was examined by HR-SEM using a Sirion scanning electron microscope (SEM, FEI Company, the Netherlands) equipped with Shottky type field emission source at 10 kV accelerating voltage. For this purpose, the samples were washed with water, dried at room temperature and sputter-coated with a Au–Pd thin film prior to HR-SEM analysis. An EDX detector was utilized to analyze film composition.The molecular weight of I was estimated using a GPC system equipped with a LKB 2150 pump, a Wyat Optilab 903 refractive index (RI) detector and a Rheodyne (Coatati, CA) injection valve with 20 μL loop (Waters Ma). Samples were eluted with THF through a set of PSS SDV Gels 100 Å and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Å columns (TOS OH Bioscience) at a flow rate of 0.8 mL min−1. The molecular weights were determined relative to polystyrene standards (Polyscience, Warrington, PA) with a molecular weight range of 500–10000. The molecular weight of III was determined using a similar set as that described above but equipped additionally with light scattering (Wyat Dawn DSP) and UV (LKB 2238 UVICORD) detectors.
000 Å columns (TOS OH Bioscience) at a flow rate of 0.8 mL min−1. The molecular weights were determined relative to polystyrene standards (Polyscience, Warrington, PA) with a molecular weight range of 500–10000. The molecular weight of III was determined using a similar set as that described above but equipped additionally with light scattering (Wyat Dawn DSP) and UV (LKB 2238 UVICORD) detectors.
1H-NMR spectra (CDCl3) were obtained by a Bruker AC 200 spectrometer in tubes with a 5 mm od. CDCl3 containing tetramethylsilane served as a solvent and shift reference, respectively.
Electrochemical measurements were conducted with an AUTOLAB PGSTAT10 potentiostat (EcoChemie, Utrecht, The Netherlands) using a single compartment three-electrode glass cell. The working electrodes were either evaporated Au on glass or polished (1200 grit emery paper) 316L stainless steel plates. The reference electrode was a saturated Hg|Hg2SO4|K2SO4(sat) electrode and a graphite rod was used as an auxiliary electrode.
Polymeric films were prepared by cyclic voltammetry (CV). The working electrode was immersed into a deaerated aqueous solution containing 0.1–0.01 M distilled pyrrole monomer and 0.45 mg mL−1 polypyrrole–PLA nanoparticle emulsion and 0.1 M NaF (used as an electrolyte to facilitate anion incorporation in the course of polymerization) at room temperature. A potential sweep between −0.8 and 1.0 V of 10 cycles and 100 mV s−1 scan rate were typically applied (unless otherwise mentioned). Then, the electropolymerized electrodes were rinsed with pure water and dried with a gentle stream of nitrogen at room temperature.
Results and discussion
Synthesis of polyglycidol-block-poly(L,L-lactide) labeled with pyrrole (III) and preparation of nanoparticles
Polyglycidol-block-poly(L,L-lactide) labeled with pyrrole (III) was synthesized by anionic copolymerization of blocked glycidol (1-ethoxyethylglycidyl ether) and L,L-lactide (Scheme 1, eqn (1)) with subsequent deblocking of hydroxyl groups in the polyglycidol block and attachment of pyrrole moieties in reaction with 3-(pyrrol-1-yl)-propanoic acid (Scheme 1, eqn (2)). The synthesis of polyglycidol-block-poly(L,L-lactide) copolymer (II) was described earlier for synthesis of poly(ethylene oxide)-block-polyglycidol–block-poly(L,L-lactide).47 The obtained poly(1-ethoxyethylglycidyl ether)-block-poly(L,L-lactide) copolymer (polyEEGly-block-PLLA, I) was characterized by GPC (see Fig. 1) and 1H NMR (see Fig. 2). The number average molecular weight (Mn) and molecular weight polydispersity (Mw/Mn) were 5700 and 1.25, respectively (relative values based on polystyrene standards calibration). Integration of these signals revealed that the number average degree of polymerization of polyEEGly and PLLA blocks was 26 and 42, respectively (see Fig. 2). Thus, the Mn of polyEEGly was 3800 and the Mn of PLLA was 3000. | ||
| Scheme 1 | ||
 | ||
| Fig. 1 GPC trace of I. | ||
 | ||
| Fig. 2 1H NMR spectrum of I. | ||
II was functionalized with 3-(pyrrole-1-yl)-propanoic acid and prepared according to a general procedure developed by Le Gall et al.46 The copolymer was characterized by GPC and 1H NMR (Fig. 3 and 4). GPC traces of eluted polymer were registered using three detectors (detector of laser light scattered at 90° (LS90), refractive index (RI) and ultraviolet light detectors). All three detectors gave a similar molecular weight distribution. A shoulder noticed at the high molecular weight side registered by the LS90 detector was probably due to the presence of some aggregates (aggregates scatter light strongly even if they are present in small number). The number average molecular weight of the copolymer determined from these GPC traces was ca. 7900.
 | ||
| Fig. 3 GPC trace of III. | ||
 | ||
| Fig. 4 1H NMR spectrum of III. | ||
Nanoparticles of this copolymer, pyrrole–PEG–PLA, were prepared using a spontaneous emulsification–solvent displacement procedure. The mean diameter particle size obtained by ALV-NIBS/HPPS was 180 nm, with particle size distribution in the range of 100–350 nm. This size range was in good correlation with a size assessment using a HR-SEM high magnification picture (×40000).
Preparation of polypyrrole–PEG–PLA nanoparticle thin film
Fig. 5 shows the cyclic voltammetry (CV) of a 0.01 M pyrrole and 0.45 mg mL−1 suspension of pyrrole–PEG–PLA nanoparticles in an aqueous solution of 0.1 M NaF using a 316L stainless steel electrode. It should be mentioned that a similar CV was obtained using an evaporated gold electrode.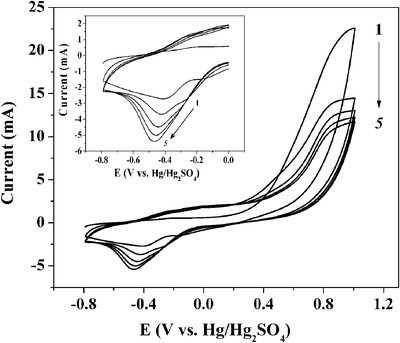 | ||
| Fig. 5 Cyclic voltammetry of a 316L stainless steel electrode in an aqueous solution containing 0.1 M NaF, 0.01 M pyrrole and 0.45 mg mL−1 pyrrole–PEG–PLA nanoparticle emulsion. Scan rate equals 100 mV s−1. | ||
Upon sweeping the potential to the positive direction in the first scan, the anodic current increased, indicating the oxidation of pyrrole monomer (Ep,a = ∼0.75 V vs. Hg|Hg2SO4|K2SO4(sat)). This anodic wave could not be detected in the absence of pyrrole, implying that the pyrrole–PEG–PLA nanoparticles do not undergo independent electrochemical polymerization and the presence of pyrrole in the solution is essential. The oxidation of the monomer occurs at more positive potentials in the successive potential scans. This shift can be attributed to the more sluggish kinetics of the monomer oxidation on the film relative to that on a bare 316L stainless steel or gold surface. During the sweep back, a small cathodic peak is observed (Ep,c = −0.45 V) which is coupled with an anodic wave (Ep,a = ∼0.0 V). The latter waves are attributed to the doping/undoping process, in which anions generally ingress and egress the film as a result of its oxidation and reduction, respectively.48–50 Since the doping/undoping wave currents depend on the thickness of the conducting polymer, they increase with each cycle provided that the film is conducting. It can be seen (Fig. 5, inset) that, indeed, the anodic currents increase during subsequent scans employing a stainless steel electrode. Yet, it should be noted that this behaviour is reversed on an evaporated gold electrode (results not shown). This different behaviour must be due to differences in the kinetics of polymerization between stainless steel and gold. The doping/undoping currents are also sensitive to the presence of water.37,39 Water is usually replaced by aprotic solvents48 or ionic liquids51 in order to enhance the electrical stability of polypyrrole. In biomedical applications, however, the presence and absorbance of water is normally unavoidable. As a matter of fact, water swelling into the polypyrrole–PEG–PLA film is necessary in order that the PLA will be able to hydrolyze and eventually biodegrade and release a drug. This will affect film conductivity. In fact, film conductivity is not an important feature for biocompatible coatings. The advantages in using a polypyrrole matrix stem mainly from its facile and cost-effective preparation and from the need to control film thickness and uniformity.
Characterization of the polypyrrole–PEG–PLA coating
Fig. 6 shows SEM images of a thin film of polylpyrrole–PEG–PLA electrodeposited on an evaporated gold electrode. After electropolymerization, the electrode was washed with water several times before conducting electron microscopy measurements. The SEM measurements reveal PEG–PLA nanoparticles embedded in the electropolymerized polypyrrole matrix (Fig. 6(a) and (b)). The images show a relatively smooth matrix in which nanoparticles are uniformly grafted without aggregation. The mean diameter particle size measured by ALV-NIBS/HPPS was ca. 180 nm, with a particle size distribution in the range of 100–350 nm, which is in agreement with the HR-SEM picture. It should be mentioned that no nanoparticles were observed either when electropolymerization was conducted in a pyrrole–PEG–PLA-free solution or in the absence of pyrrole (not shown). The latter inspection strengthens the electrochemical observation described before, that no electropolymerization takes place when pyrrole monomer is absent. | ||
| Fig. 6 SEM images of the film synthesized in the same conditions as in Fig. 5, but on an evaporated gold electrode. Magnifications: (a) ×5000, marker equals 5 μm, (b) ×40000, marker equals 500 nm. | ||
Fig. 7 presents the morphological changes inside the polypyrrole–PEG–PLA nanocomposite film caused by immersion in pure acetonitrile (ACN) solution. Small voids in the PEG–PLA nanoparticles, which could not be found in the non-immersed film, were detected after one hour of immersion. These voids are caused by the dissolution of PLA by ACN. It is well-known that PLA particles are susceptible to hydrolysis. Elemental analysis of the polypyrrole–PEG–PLA matrix was also used to confirm the presence of PEG–PLA nanoparticles and showed that the oxygen to carbon weight ratio in the particles (21.22) is larger than in the polypyrrole matrix (16.00). This is expected, owing to the oxygen that constitutes the PEG–PLA building blocks and is absent in the conductive matrix. This result is in line with the solubility of the nanoparticles in ACN. In the case of the polypyrrole matrix, fluoride was also detected due to the ingress of fluoride in the course of the electropolymerization.
 | ||
| Fig. 7 SEM images of the film shown in Fig. 6 after immersion in ACN for one hour. Magnifications: (a) ×10000, marker equals 2 μm, (b) ×68300, marker equals 500 nm. | ||
Conclusions
Polypyrrole–PEG–PLA nanocomposite film was deposited on gold and stainless steel surfaces by electropolymerization of pyrrole and pyrrole-functionalized PEG–PLA nanoparticles. This novel approach comprises the advantages of electropolymerization, which allows the fine control of an organic coating, and the nanoparticulate nature of PLA, which leads to the electrodeposition of nanoparticles. Taking into account the biodegradable nature of the PLA nanoparticles and the polymer matrix, this polypyrrole–PEG–PLA film could therefore be considered as a feasible partially biodegradable electrodeposited nanocomposite coating for metal implants. The biocompatibility of this novel coating is currently under examination.Acknowledgements
This work was supported by the Hebrew University through an applied grant and by Elutex. The unit for nanocharacterization of the Hebrew University is acknowledged. This work was supported in part by a grant from the Israel Science Foundation (grant 200/02). M. Gadzinowski and S. Slomkowski would like to acknowledge support of State Committee of Scientific Research, grant No. BZ-KBN-070/T09/2001/3.References
- W. A. Gazotti, V. F. Juliano and M. De Paoli, Polym. Degrad. Stab., 1993, 42, 317 CrossRef.
- B. A. Netomarco and M. A. De Paoli, Polym. Degrad. Stab., 1993, 40, 59 CrossRef.
- L. Cen, K. G. Neoh and E. T. Kang, Biosens. Bioelectron., 2003, 18, 363 CrossRef CAS.
- M. Gao, l. Dai and G. G. Wallace, Synth. Met., 2003, 137, 1393 CrossRef CAS.
- Y. Xian, Y. Hu, F. Liu, Y. Xian, H. Wang and L. Jin, Biosens. Bioelectron., 2006, 21, 1996 CrossRef CAS.
- K. Arora, A. Chaubey, R. Singhal, R. P. Singh, M. K. Pandey, S. B. Samanta, B. D. Malhotra and S. Chand, Biosens. Bioelectron., 2006, 21, 1777 CrossRef CAS.
- Rajesh, S. S. Pandey, W. Takashima and K. Kaneto, Curr. Appl. Phys., 2005, 5, 184 Search PubMed.
- J. Wang and M. Musameh, Anal. Chim. Acta, 2005, 539, 209 CrossRef CAS.
- Rajesh, S. S. Pandey, W. Takashima and K. Kaneto, J. Appl. Polym. Sci., 2004, 93, 927 CrossRef.
- S. S. Razola, B. L. Ruiz, N. M. Diez, H. B. Mark and J. M. Kauffmann, Biosens. Bioelectron., 2002, 17, 921 CrossRef.
- G. Bidan, C. Lopez, F. Mendes-Viegas and A. Gadelle, Biosens. Bioelectron., 1995, 10, 219 CrossRef CAS.
- L. L. Miller and Q.-X. Zhou, Macromolecules, 1987, 20, 1594 CrossRef CAS.
- L. L. Miller, B. Zinger and Q. X. Zhou, J. Am. Chem. Soc., 1987, 109, 2267 CrossRef CAS.
- Q.-X. Zhou, L. Miller and J. Valentine, J. Electroanal. Chem., 1989, 261, 147 CrossRef CAS.
- Y. Li, X. Y. Cheng, M. Y. Leung, J. Tsang, X. M. Tao and M. C. W. Yuen, J. Chem. Soc., Chem. Commun., 1992, 827 RSC.
- H.-T. Lee and Y.-C. Liu, Polymer, 2005, 46, 10727 CrossRef CAS.
- S. J. Tian, J. Liu, T. Zhu and W. Knoll, Chem. Mater., 2004, 16, 4103 CrossRef CAS.
- X. J. Zhou, A. J. Harmer, N. F. Heinig and K. T. Leung, Langmuir, 2004, 20, 5109 CrossRef CAS.
- A. Houdayer, R. Schneider, D. Billau, J. Ghanbaja and J. Lambert, Synth. Met., 2005, 151, 165 CrossRef CAS.
- L. Z. Zheng and J. H. Li, J. Electroanal. Chem., 2005, 577, 137 CrossRef CAS.
- P. G. Nicholson, V. Ruiz and J. V. Macpherson, Chem. Commun., 2005, 1052 RSC.
- P. J. Kulesza, M. Chojak and K. Karnicka, Chem. Mater., 2004, 16, 4128 CrossRef CAS.
- J. E. Park, S. G. Park and A. Koukitu, Synth. Met., 2004, 141, 265 CrossRef CAS.
- L. Zhai and R. D. Mccullough, J. Mater. Chem., 2004, 14, 141 RSC.
- A. P. O’Mullane, S. E. Dale and J. V. Macpherson, Chem. Commun., 2004, 1606 RSC.
- S. J. Tian, J. Y. Liu and T. Zhu, Chem. Commun., 2003, 2738 RSC.
- M. Lee, B. W. Kim and J. D. Nam, Mol. Cryst. Liq. Cryst., 2003, 407, 397 CAS.
- H. S. Kim, B. H. Sohn and W. Lee, Thin Solid Films, 2002, 419, 173 CrossRef CAS.
- N. Cioffi, L. Torsi and I. Losito, Electrochim. Acta, 2001, 46, 4205 CrossRef CAS.
- M. A. Breimer, G. Yevgeny and S. Sy, Nano Lett., 2001, 1, 305 CrossRef CAS.
- R. Gangopadhyay and A. De, Chem. Mater., 2000, 12, 608 CrossRef CAS.
- K. V. Sarathy and K. S. Narayan, Curr. Sci., 1999, 77, 678 CAS.
- X. Zou, H. Bao, H. Guo, L. Zhang, L. Qi, J. Jiang, L. Niu and S. Dong, J. Colloid Interface Sci., 2006, 295, 401 CrossRef CAS.
- (a) S. S. Ran and M. Biswas, Synth. Met., 2000, 108, 231 CrossRef; (b) Y. Xian, Y. Hu, F. Liu, Y. Xian, H. Wang and L. Jin, Biosens. Bioelectron., 2006, 21, 1996 CrossRef CAS.
- A. Akelah, N. El-Deen, A. Hiltner, E. Baer and A. Moet, Mater. Lett., 1995, 22, 97 CrossRef CAS.
- S. H. Jang, M. G. Han and S. S. Im, Synth. Met., 2000, 110, 17 CrossRef CAS.
- G. Shi, M. Rouabhia, Z. Wang, L. H. Dao and Z. Zhang, Biomaterials, 2004, 25, 2477 CrossRef CAS.
- P. R. Supronowicz, P. M. Ajayan, K. R. Ullmann, B. P. Arulanandam, D. W. Metzger and R. Bizious, J. Biomed. Mater. Res., 2002, 59, 499 CrossRef CAS.
- Z. X. Wang, C. Roberge, Y. Wan, L. H. Dao, R. Guidoin and Z. Zhang, J. Biomed. Mater. Res., Part A, 2003, 66, 738 Search PubMed.
- J. M. Anderson and M. S. Shive, Adv. Drug Delivery Rev., 1997, 28, 5 CrossRef CAS.
- L. A. Guzman, V. Labhasetwar, C. Song, Y. Jang, A. M. Lincoff, R. Levy and E. J. Topol, Circulation, 1996, 94, 1441 CAS.
- I. Fishbein, M. Chorny, L. Rabinovich, S. Banai, I. Gati and G. Golomb, J. Controlled Release, 2000, 65, 221 CrossRef CAS.
- R. Gref, A. Domb, P. Quellac, T. Blunk, R. H. Muller, J. M. Verbavatz and R. Langer, Adv. Drug Delivery Rev., 1995, 16, 215 CrossRef CAS.
- D. Williams, A. Dash, D. Leslie-pelecky and V. Labhasetwar, J. Pharm. Sci., 2004, 93, 1804 CrossRef.
- A. O. Fitton, J. Hill, D. E. Jane and R. Miller, Synthesis, 1987, 12, 1140 CrossRef.
- T. Le Gall, M. S. Passos, S. K. Ibrahim, S. Morlat-Theiras, C. Sudbrake, S. A. Fairhurst, M. A. Queiros and Ch. J. Pickett, J. Chem. Soc., Perkin Trans. 1, 1999, 1657 RSC.
- M. Gadzinowski and S. Sosnowski, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3750 CrossRef CAS.
- T. J. Skotheim, in Handbook of Conducting Polymers, Marcel Dekker, New York, 1986. pp. 81–117 Search PubMed.
- A. F. Diaz, J. I. Castillo, J. A. Logan and W. Y. Lee, J. Electroanal. Chem., 1981, 129, 115 CrossRef CAS.
- E. T. Kang, T. C. Tan and K. G. Neoh, Polymer, 1986, 27, 1958 CrossRef CAS.
- W. Lu, A. G. Fadeev, B. Qi, E. Smela, B. R. Mattes, J. Ding, G. M. Spinks, J. Mazurkiewicz, D. Zhou, G. G. Wallace, D. R. MacFarlane, S. A. Forsyth and M. Forsyth, Science, 2002, 297, 983 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2007 |