Synthesis and characterisation of peripherally functionalised dendritic molecules
Sarah J.
Atkinson
,
Vicky-June
Ellis
,
Sue E.
Boyd
and
Christopher L.
Brown
*
Chemical Biology Program, Eskitis Institute, Griffith University, Nathan, Brisbane, Australia. E-mail: c.l.brown@griffith.edu.au; Fax: +61-7-37357656; Tel: +61-7-37357293
First published on 13th October 2006
Abstract
A number of peripherally functionalised dendritic molecules were synthesised in almost quantitative yield by a synthetic method involving the reaction between amines and isocyanates. The peripheral functional groups were incorporated by preparing a number of branched subunits, based on tris(hydroxymethyl)aminomethane (TRIS), 1, possessing three nitro, methoxy, methyl or maleimide terminal functionalities. Attachment of these branched units to the core molecules 4,4′-methylenebis(phenyl isocyanate)13 and 1,3,5-benzene triisocyanate20 afforded the corresponding dendritic molecules possessing 6 or 9 peripheral functional groups. Functional group conversions on the dendritic molecules have been successfully carried out, including hydrogenation of the terminal nitro to the corresponding amine and cleavage of the methoxy ether to give the corresponding phenol.
Introduction
A rising demand for well-defined, functional materials for nano-scale applications has stimulated the development of synthetic procedures for the efficient assembly of dendritic and polymeric species.1 Several synthetic approaches to these systems feature architectural control while still maintaining flexibility in the incorporation of functional groups.2 However, many approaches to dendrimer syntheses, particularly in later generations, require high monomer loading and involve prolonged, repetitive, chromatographic separations, generating considerable waste.3 In an attempt to overcome this problem a growing number of synthetic approaches are utilising facile coupling techniques for the assembly of macromolecular dendritic and polymeric structures.4 Recently, several urea-based syntheses have been reported for the divergent growth of dendrimers5–7 and other macro-molecular structures.8 Here we report a highly efficient, facile approach for the modular synthesis of a diverse range of peripherally functionalised structures that can serve as scaffolds for the assembly of more complex macromolecular structures for use in nanotechnology, biological and materials sciences. The syntheses utilise the well-documented reaction between an isocyanate and an amine and afford peripherally functionalised nanoparticles in excellent yields and purity.Results and discussion
Synthetic strategy
The synthetic strategy utilised in this body of work was based on the preparation of a number of branched synthetic building blocks possessing differing terminal functionality. These dendrons were then attached to bi- and tri-functional core molecules via urea linkages, by utilising the facile reaction occurring between amines and isocyanates. Such a modular strategy provides ample opportunity for tailoring of the final molecules for specific applications and properties.Synthesis of branched dendrons with peripheral functionality
The synthesis of the branched dendrons, bearing three terminal residues and based on the inexpensive starting material tris(hydroxymethyl)aminomethane (TRIS) 1, involved the coupling of 3.3 equivalents of an appropriately para-substituted benzoic acid chloride with Boc protected TRIS2 in dichloromethane (DCM) in the presence of triethylamine (Scheme 1). | ||
| Scheme 1 Synthesis of the branched dendrons. | ||
The p-nitro, p-methoxy and p-methyl benzoyl chloride starting materials were obtained commercially, whilst N-(4-carboxyphenyl)maleimide was synthesised according to literature procedures.9 Conversion of N-(4-carboxyphenyl)maleimide to the required acid chloride was carried out immediately prior to use via standard procedures using thionyl chloride. Initial synthesis of the maleimide 3-mer dendron (N-(tert-butyloxycarbonyl)-1,1,1-tris(4-maleimidobenzoyloxy-methyl)methylamine) 6 resulted in low yields due to unwanted polymerisation. Subsequent procedures utilising tert-butylcatechol to suppress polymerisation9 resulted in higher yields being obtained. The Boc protected dendrons 3–6 were synthesised in yields ranging from 41–92%. Deprotection of the Boc protected dendrons 3 and 4 in DCM with trifluoroacetic acid (TFA), followed by the addition of 1 M sodium carbonate afforded the TFA salts 7 and 9, respectively. The corresponding free base amines 8 and 10 were obtained on further treatment of the TFA salts with sodium carbonate. Deprotection of the Boc protected dendrons 5 and 6 afforded the free amines 11 and 12 directly after treatment with sodium carbonate.
Synthesis of functionalised branched molecules containing 6- and 9-peripheral functionalities
The modular strategy devised for the synthesis of branched molecules containing 6- and 9-peripheral functionalities provides for flexibility in the level of complexity available for elaboration, as well as increasing the variety of peripheral functional groups that could be incorporated. Initial investigations were carried out on the commercially available bi-functional 4,4′-methylenebis(phenyl isocyanate)13 as the core unit. The synthetic methodology for the assembly of these linear 6-mer systems is shown in Scheme 2. | ||
| Scheme 2 Synthesis of the 6-mer macromolecules. | ||
The success of this system can be best illustrated through the resultant NMR spectra of the ‘crude’ 6-nitro species as seen in Fig. 1. The symmetry of the fully functionalised dendritic system is evidenced in the NMR spectrum. Less symmetrical species, suggesting incomplete reactions, were not observed in the crude products isolated in all cases. Formation of the urea linkages were confirmed through two-dimensional NMR experiments and the corresponding cross peaks can be clearly identified in the gHMBC spectrum (Fig. 2).
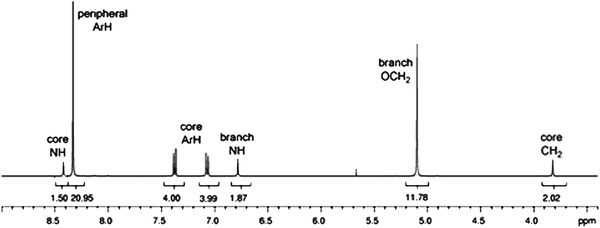 | ||
| Fig. 1 1H NMR spectrum (400 MHz, 298 K, CD3COCD3–CD3SOCD3) of the crude 6-mer, 14. | ||
 | ||
| Fig. 2 Expansion of the 1H/13C gHMBC spectrum of the 6-mer, 14 (400 MHz, 298 K, CD3COCD3–CD3SOCD3). Basic 1H and 13C{1H} spectra are provided as axes references. Crosspeaks correlating the NH protons and the carbon of the central carbonyl of the urea linkage are highlighted. | ||
1,3,5-Benzene triisocyanate 20 was selected as the tri-functional core unit for the synthesis of the more radially distributed 9-mer systems,. This material was prepared, according to a literature procedure,7 from the corresponding 1,3,5-benzenetricarbonyl triazide precursor 19, via a Curtius rearrangement. Once prepared the acylazide precursor 19 was stored at –20 °C until required, as this compound is reported to be explosive under heat or pressure in the solid state.8 The synthetic methodology for the assembly of the 9-mer systems is shown in Scheme 3.
 | ||
| Scheme 3 Synthesis of the 9-mer branched molecules. | ||
In most cases the products precipitated from the reaction mixture after 18 h and were isolated simply by filtration. 1H NMR analysis of the reaction products for the formation of the nitro 6-mer 14 (Scheme 2) showed the formation of spectroscopically pure product in a 75% yield. However, the use of the trifunctional core 20 to assemble the analogous 9-mer nitro species 21 resulted in poor yields. Further investigation found that the application of gentle heat to the reaction afforded required compound 21 in a 92% yield (Scheme 3). Consequently, subsequent reactions to construct the 6-mer and 9-mer species were carried out in refluxing DCM. Formation of the methoxy 15 and maleimido 16 functionalised 6-mers recorded yields of 74 and 94% (Scheme 2), whilst the construction of the methoxy 22, methyl 23 and maleimide 24 terminated 9-mers achieved yields of 96, 97, and 99%, respectively (Scheme 3). Solubility of both the 6-mer and 9-mer species varies greatly with varying external functionality. In all cases the methoxy and maleimido functionalised molecules were soluble in dichloromethane and were isolated via solvent removal at reduced pressure.
Conversion of the peripheral nitro functionalised species 14 and 21 to the corresponding amines occurred smoothly via hydrogenation using 5% Pd/C under elevated temperatures and pressure (DMF, 55 °C, 600 psi) and afforded yields of the polyamine 6-mer 17 in 62% yield and the 9-mer 25 in 60% yield, respectively. Similarly, conversion of the methoxy protected 9-mer 22, to the phenolic compound 26 (AlBr3, dodecane thiol, DCM) afforded the 9-mer polyphenol 26 in 87% yield.
Conclusions
We have demonstrated the facile assembly of highly monodisperse branched molecules possessing a range of peripheral functionalities, based upon the high yielding coupling reaction occurring between amines and isocyanates. The uniform attachment of the dendrons to the bi- and tri-functionalised cores is highly efficient and yields compounds of high purity due to the quantitative reaction between the amines on the branched dendrons and the isocyanates present on the core molecules. Our investigations are continuing into the application of these molecules as a basis for the assembly of more complex macromolecular structures for applications in biological and materials science.Experimental
General methods and materials
Anhydrous solvents were prepared according to literature methods.10 Most reagents and starting materials were obtained from commercial sources (Sigma-Aldrich) and used without further purification. 4-Maleimidobenzoic acid9 and 1,3,5-benzenetricarbonyl triazide719 were prepared according to published procedures. All reactions were performed in flame-dried or oven-dried glassware under an inert atmosphere of nitrogen. In all cases the isocyanate was prepared immediately prior to coupling with the appropriate amine. Analytical TLC was carried out on Merck aluminium TLC plates coated with silica gel 60 F254 (0.2 mm) and visualised by ultraviolet light (254 nm) or in an iodine vapour tank. Chromatographic separations were carried out by column chromatography with Merck silica gel 60 Å (230–400 mesh) as the stationary phase.NMR spectra were recorded on a Varian Unity INOVA spectrometer. 1H (400 MHz) and 13C (100 MHz) chemical shifts were referenced to the residual solvent peaks. gCOSY, gHSQC and gHMBC spectra were acquired using the standard sequences contained in the VNMR6.1C software package. FTIR spectra were recorded in the range of 4000–400 cm–1 on a Thermo Nicolet-Nexus FTIR spectrometer. Melting points were measured using the capillary method on a GallenKamp Variable Temperature Apparatus and are uncorrected. Mass spectra were recorded on a Fisons VG-Platform II spectrometer, using positive and negative mode electrospray as the ionisation technique and Mass Lynx Version I (IBM) software to acquire and process ESMS data. Lithium was added to ESMS scans as LiBr. HRMS were carried out by the Mass Spectrometry Facility, Griffith University on a Bruker Daltonics Apex III 4.7e Fourier Transform Mass Spectrometer, fitted with an Apollo API source and are within 2.5 ppm. Elemental analyses were carried out by the Microanalytical Service, Department of Chemistry at the University of Queensland, Brisbane.
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 77.83 (Cq–CH3), 60.45 (CH2), 60.24 (Cq–CH2), 28.22 (CH3).
O), 150.30 (ArC–NO2), 134.68 (ArC–CO2H), 130.74 (ArC–H), 123.76 (ArC–H), 78.79 (Cq–CH3), 63.78 (CH2), 56.57 (Cq–CH2), 28.01 (C–CH3). Anal. calcd for C30H28N4O14: C, 53.89; H, 4.22; N, 8.38. Found: C, 54.12; H, 4.35; N, 8.23%.
O), 131.37 (ArC–H), 121.53 (ArC–CO2R), 113.93 (ArC–H), 78.47 (Cq–CH3), 62.85 (CH2), 56.77 (Cq–CH2), 55.48 (O–CH3), 28.04 (C–CH3). Anal. calcd for C33H37NO11: C, 63.55; H, 5.98; N, 2.25. Found: C, 63.64; H, 6.01; N, 2.21%.
O), 143.78 (ArC–CH3), 129.29 (ArC–H), 129.20 (ArC–H), 126.61 (ArC–CO2R), 78.49 (Cq–CH3), 62.96 (CH2), 56.73 (Cq–CH2), 28.03 (C–CH3), 21.13 (ArC–CH3). Anal. calcd for C33H37NO8: C, 68.85; H, 6.48; N, 2.43. Found: C, 69.12; H, 6.46; N, 2.42%.
CH), 4.76 (s, 6H, CH2), 1.36 (s, 9H, CH3). 13C NMR (100 MHz, DMSO-d6) δ 169.43 (CONCO), 164.66 (CO2R), 154.71 (NH–CO), 135.95 (ArC–NR), 134.86 (RHCCHR), 129.90 (ArC–H), 128.02 (ArC–CO2R), 126.19 (ArC–H), 78.57 (Cq–CH3), 63.31 (CH2), 56.75 (Cq–CH2), 28.05 (C–CH3). Anal. calcd for C42H34N4O14: C, 61.61; H, 4.19; N, 6.84. Found: C, 61.76; H, 4.24; N, 6.87%.
CH), 4.63 (s, 6H, CH2). 13C NMR (100 MHz, acetone-d6) δ 170.19 (CONCO), 165.99 (CO2R), 137.30 (ArC–NR), 135.61 (RHCCHR), 131.02 (ArC–H), 129.59 (ArC–CO2R), 126.80 (ArC–H), 68.10 (CH2), 55.65 (Cq–CH2). Anal. calcd for C37H26N4O12: C, 61.84; H, 3.65; N, 7.80. Found: C, 61.62; H, 3.74; N, 7.59%.
ONH), 150.28 (ArC–NO2), 137.55 (core-ArC–NHR), 134.82 (core-ArC–H),134.55 (ArC–CO2R),130.77 (p-NO2-ArC–H), 128.8 (core-ArC–CH2), 123.73 (p-NO2-ArC–H), 118.14 (core-ArC–H), 64.51(CH2), 56.65 (Cq), 39.51 coincident with residual solvent (core-CH2). Anal. calcd for C65H50N10O26: C, 56.28; H, 3.63; N, 10.10. Found: C, 56.30; H, 3.65; N, 9.85%.
ONH), 137.68 (core-ArC–NHR), 134.67 (ArC–CO2R), 131.39 (core-ArC–H), 128.80 (core-ArC–CH2), 121.37 (p-CH3O–ArC–H), 118.02 (core-ArC–NHR), 113.95 (p-CH3O–ArC–H), 63.62 (CH2), 56.79 (Cq), 55.47 (CH3), 39.51 (core-CH2). Anal. calcd for C71H68N4O20: C, 65.73; H, 5.28; N, 4.32. Found: C, 65.27; H, 5.25; N, 4.03%.
CH), 6.69 (br. s, 2H, NH), 4.87 (s,12H, CH2), 3.75 (br. s, 2H, core-CH2). 13C NMR(100 Mhz, DMSO-d6) δ 169.40 (OCNCO), 164.70 (CO2R), 154.72 (NHCONH), 137.61 (core-ArC–H), 135.95 (ArC–NR2), 134.83 (RHCCHR), 134.73 (ArC–CO2R), 129.91 (p-maleimide-ArC–H), 128.83 (core-ArC–CH2), 127.88 (core-ArC–H), 126.15 (p-maleimide-ArC–H), 118.19 (core-ArC–NHR), 64.16 (CH2), 56.76 (Cq), 39.51 coincident with residual solvent (core-CH2). Anal. calcd for C89H62N10O26: C, 63.35; H, 3.70; N, 8.30. Found: C, 62.46; H, 3.72; N, 7.96%.
ONH), 153.71 (ArC–NH2), 137.74 (core-ArC–H), 134.54 (ArC–CO2), 131.28 (core-ArC–H), 128.83 (core-ArC–CH2), 117.96 (core-ArC–NHR), 115.21 (p-NH2–ArC–H), 112.59 (p-NH2–ArC–H), 62.88 (CH2), 56.94 (Cq), 39.51 coincident with residual solvent (core-CH2).
ONH), 151.70 (ArC–NO2), 141.47 (ArC–NHR), 135.96 (ArC–CO2R), 131.83 (p-NO2–ArC–H), 124.51 (p-NO2–ArC–H), 103.43 (core-ArC–H), 65.73 (CH2), 58.50 (Cq).
ONH), 141.70 (ArC–NHR), 132.55 (p-CH3O–ArC–H), 123.02 (ArC–CO2R), 114.73 (p-CH3O–ArC–H), 102.56 (core-ArC–H), 64.87 (CH2), 58.57 (Cq), 56.01 (CH3).
ONH), 144.90 (ArC–CH3), 141.64 (ArC–NHR), 130.52 (p-CH3–ArC–H), 130.10 (p-CH3-ArC–H), 128.08 (ArC–CO2R), 102.57 (core-ArC–H), 64.94 (CH2), 58.53 (Cq), 21.66 (CH3).
CH), 6.53 (br. s, 3H, NH), 4.87 (s, 18H, CH2). 3C NMR (100 MHz, DMSO-d6) δ 169.4 (CONCO), 164.69 (CO2R), 154.55 (NHCONH), 140.43 (core-ArC–NHR), 135.96 (ArC–NR2), 134.83 (RHCCHR), 129.93 (p-maleimide-ArC–H), 127.85 (ArC–CO2R), 126.15 (p-maleimide-ArC–H), 100.57 (core-ArC–H), 64.20 (CH2), 56.70 (Cq). Anal. calcd for C120H81N15O39.H2O: C, 60.69; H, 3.52; N, 8.85. Found: C, 60.43; H, 3.71; N, 8.71%.
ONH), 153.70 (ArC–NH2), 140.51 (core-ArC–NHR), 131.29 (pNH2–ArC–H), 115.18 (pNH2–ArC–H), 112.62 (core-ArC–H), 62.89 (CH2), 56.87 (Cq).
ONH), 140.50 (core-ArC–NHR), 131.66 (p-OH-ArC–H), 119.80 (ArC–CO2R), 115.35 (pOH–ArC–H), 100.15 (core-ArC–H), 63.51 (CH2), 56.76 (Cq).
O), 77.83 (Cq–CH3), 60.45 (CH2), 60.24 (Cq–CH2), 28.22 (CH3).
O), 150.30 (ArC–NO2), 134.68 (ArC–CO2H), 130.74 (ArC–H), 123.76 (ArC–H), 78.79 (Cq–CH3), 63.78 (CH2), 56.57 (Cq–CH2), 28.01 (C–CH3). Anal. calcd for C30H28N4O14: C, 53.89; H, 4.22; N, 8.38. Found: C, 54.12; H, 4.35; N, 8.23%.
O), 131.37 (ArC–H), 121.53 (ArC–CO2R), 113.93 (ArC–H), 78.47 (Cq–CH3), 62.85 (CH2), 56.77 (Cq–CH2), 55.48 (O–CH3), 28.04 (C–CH3). Anal. calcd for C33H37NO11: C, 63.55; H, 5.98; N, 2.25. Found: C, 63.64; H, 6.01; N, 2.21%.
O), 143.78 (ArC–CH3), 129.29 (ArC–H), 129.20 (ArC–H), 126.61 (ArC–CO2R), 78.49 (Cq–CH3), 62.96 (CH2), 56.73 (Cq–CH2), 28.03 (C–CH3), 21.13 (ArC–CH3). Anal. calcd for C33H37NO8: C, 68.85; H, 6.48; N, 2.43. Found: C, 69.12; H, 6.46; N, 2.42%.
CH), 4.76 (s, 6H, CH2), 1.36 (s, 9H, CH3). 13C NMR (100 MHz, DMSO-d6) δ 169.43 (CONCO), 164.66 (CO2R), 154.71 (NH–CO), 135.95 (ArC–NR), 134.86 (RHCCHR), 129.90 (ArC–H), 128.02 (ArC–CO2R), 126.19 (ArC–H), 78.57 (Cq–CH3), 63.31 (CH2), 56.75 (Cq–CH2), 28.05 (C–CH3). Anal. calcd for C42H34N4O14: C, 61.61; H, 4.19; N, 6.84. Found: C, 61.76; H, 4.24; N, 6.87%.
CH), 4.63 (s, 6H, CH2). 13C NMR (100 MHz, acetone-d6) δ 170.19 (CONCO), 165.99 (CO2R), 137.30 (ArC–NR), 135.61 (RHCCHR), 131.02 (ArC–H), 129.59 (ArC–CO2R), 126.80 (ArC–H), 68.10 (CH2), 55.65 (Cq–CH2). Anal. calcd for C37H26N4O12: C, 61.84; H, 3.65; N, 7.80. Found: C, 61.62; H, 3.74; N, 7.59%.
ONH), 150.28 (ArC–NO2), 137.55 (core-ArC–NHR), 134.82 (core-ArC–H),134.55 (ArC–CO2R),130.77 (p-NO2-ArC–H), 128.8 (core-ArC–CH2), 123.73 (p-NO2-ArC–H), 118.14 (core-ArC–H), 64.51(CH2), 56.65 (Cq), 39.51 coincident with residual solvent (core-CH2). Anal. calcd for C65H50N10O26: C, 56.28; H, 3.63; N, 10.10. Found: C, 56.30; H, 3.65; N, 9.85%.
ONH), 137.68 (core-ArC–NHR), 134.67 (ArC–CO2R), 131.39 (core-ArC–H), 128.80 (core-ArC–CH2), 121.37 (p-CH3O–ArC–H), 118.02 (core-ArC–NHR), 113.95 (p-CH3O–ArC–H), 63.62 (CH2), 56.79 (Cq), 55.47 (CH3), 39.51 (core-CH2). Anal. calcd for C71H68N4O20: C, 65.73; H, 5.28; N, 4.32. Found: C, 65.27; H, 5.25; N, 4.03%.
CH), 6.69 (br. s, 2H, NH), 4.87 (s,12H, CH2), 3.75 (br. s, 2H, core-CH2). 13C NMR(100 Mhz, DMSO-d6) δ 169.40 (OCNCO), 164.70 (CO2R), 154.72 (NHCONH), 137.61 (core-ArC–H), 135.95 (ArC–NR2), 134.83 (RHCCHR), 134.73 (ArC–CO2R), 129.91 (p-maleimide-ArC–H), 128.83 (core-ArC–CH2), 127.88 (core-ArC–H), 126.15 (p-maleimide-ArC–H), 118.19 (core-ArC–NHR), 64.16 (CH2), 56.76 (Cq), 39.51 coincident with residual solvent (core-CH2). Anal. calcd for C89H62N10O26: C, 63.35; H, 3.70; N, 8.30. Found: C, 62.46; H, 3.72; N, 7.96%.
ONH), 153.71 (ArC–NH2), 137.74 (core-ArC–H), 134.54 (ArC–CO2), 131.28 (core-ArC–H), 128.83 (core-ArC–CH2), 117.96 (core-ArC–NHR), 115.21 (p-NH2–ArC–H), 112.59 (p-NH2–ArC–H), 62.88 (CH2), 56.94 (Cq), 39.51 coincident with residual solvent (core-CH2).
ONH), 151.70 (ArC–NO2), 141.47 (ArC–NHR), 135.96 (ArC–CO2R), 131.83 (p-NO2–ArC–H), 124.51 (p-NO2–ArC–H), 103.43 (core-ArC–H), 65.73 (CH2), 58.50 (Cq).
ONH), 141.70 (ArC–NHR), 132.55 (p-CH3O–ArC–H), 123.02 (ArC–CO2R), 114.73 (p-CH3O–ArC–H), 102.56 (core-ArC–H), 64.87 (CH2), 58.57 (Cq), 56.01 (CH3).
ONH), 144.90 (ArC–CH3), 141.64 (ArC–NHR), 130.52 (p-CH3–ArC–H), 130.10 (p-CH3-ArC–H), 128.08 (ArC–CO2R), 102.57 (core-ArC–H), 64.94 (CH2), 58.53 (Cq), 21.66 (CH3).
CH), 6.53 (br. s, 3H, NH), 4.87 (s, 18H, CH2). 3C NMR (100 MHz, DMSO-d6) δ 169.4 (CONCO), 164.69 (CO2R), 154.55 (NHCONH), 140.43 (core-ArC–NHR), 135.96 (ArC–NR2), 134.83 (RHCCHR), 129.93 (p-maleimide-ArC–H), 127.85 (ArC–CO2R), 126.15 (p-maleimide-ArC–H), 100.57 (core-ArC–H), 64.20 (CH2), 56.70 (Cq). Anal. calcd for C120H81N15O39.H2O: C, 60.69; H, 3.52; N, 8.85. Found: C, 60.43; H, 3.71; N, 8.71%.
ONH), 153.70 (ArC–NH2), 140.51 (core-ArC–NHR), 131.29 (pNH2–ArC–H), 115.18 (pNH2–ArC–H), 112.62 (core-ArC–H), 62.89 (CH2), 56.87 (Cq).
ONH), 140.50 (core-ArC–NHR), 131.66 (p-OH-ArC–H), 119.80 (ArC–CO2R), 115.35 (pOH–ArC–H), 100.15 (core-ArC–H), 63.51 (CH2), 56.76 (Cq).
References
- (a) Dendrimers and Dendrons: Concepts, Syntheses, Applications, ed. G. R. Newkome, C. N. Moorefield and F. Vögtle, Wiley-VCH, Weinheim, 2001 Search PubMed; (b) Dendrimers and Other Dendritic Polymers, ed. J. M. J. Fréchet and D. A. Tomalia, John Wiley & Sons, Chichester, 2001 Search PubMed; (c) C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS.
- A. W. Bosman, R. Vestverg, A. Heumann, J. M. J. Frechet and C. J. Hawker, J. Am. Chem. Soc., 2003, 125, 715–728 CrossRef CAS.
- P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Frechet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928–3932 CrossRef CAS.
- (a) P. Wu, M. Malkoch, J. N. Hunt, R. Vestberg, E. Kaltgrad, M. G. Finn, V. V. Fokin, K. B. Sharpless and C. J. Hawker, Chem. Commun., 2005, 46, 5775 RSC; (b) J. W. Lee, J. H. Kim and B.-K. Kim, Tetrahedron Lett., 2006, 47, 2683–2686 CrossRef CAS; (c) J. W. Lee, J. H. Kim, B.-K. Kim, J. Hee, S. C. Han, W. S. Shin and S.-H. Jin, Macromolecules, 2006, 39, 2418–2422 CrossRef CAS; (d) M. J. Joralemon, R. K. O’Reilly, J. B. Matson, A. K. Nugent, C. J. Hawker and K. L. Wooley, Macromolecules, 2005, 38, 5436–5443 CrossRef CAS; (e) R. K. O’Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2005, 46, 92–93 CAS; (f) B. Helms, J. L. Mynar, C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 2004, 126, 15020–15021 CrossRef CAS; (g) P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Frechet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928–32 CrossRef CAS.
- M. Okaniwa, K. Takeuchi, M. Asai and M. Ueda, Macromolecules, 2002, 35, 6224–6231 CrossRef CAS.
- M. Okaniwa, K. Takeuchi, M. Asai and M. Ueda, Macromolecules, 2002, 35, 6232–6238 CrossRef CAS.
- A. V. Ambade and A. J. Kumar, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1295–1304 CrossRef CAS.
- J. J. vanGorp, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759–14769 CrossRef CAS.
- T. Oishi and M. Fujimoto, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 1821–1830 CrossRef CAS.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Permagon Press, Oxford, 3rd edn., 1988 Search PubMed.
- J.-H. Kang, H.-E. Chung, S. Y. Kim, Y. Kim, J. Lee, N. E. Lewin, L. V. Pearce, P. M. Blumberg and V. E. Marquez, Bioorg. Med. Chem., 2003, 11, 2529–2539 CrossRef CAS.
- M. Hasan, D. Bethell and M. Brust, J. Am. Chem. Soc., 2002, 124, 1132–1133 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2007 |