Control of macromolecular structure and function using covalently attached double-stranded DNA constraints†
Scott K.
Silverman
*
Department of Chemistry, University of Illinois at Urbana-Champaign, 600 South Mathews Avenue, Urbana, Illinois 61801, USA
First published on 14th November 2006
Abstract
The biophysical properties of DNA suggest its use for applications beyond serving as the genetic material. Several recent reports describe the use of covalently attached double-stranded DNA for controlling the structures of other macromolecules such as protein and RNA. These exploitations of DNA rigidity are conceptually distinct from many other studies in the area of “DNA nanotechnology”. Double-stranded DNA constraints provide a means of introducing selective tension onto other molecules. This should facilitate fundamental investigations of macromolecular folding landscapes and tertiary interactions, as well as allow study of the mechanotransduction of biochemical signals. Use of a DNA constraint as the key element of a sensor has already been demonstrated, and such applications will be enhanced by improvements in the signal readout methods. If practical challenges such as delivery and stability can be addressed, these new efforts may also enable development of selective sensors for in vivo applications.
 Scott K. Silverman | Scott K. Silverman grew up in Los Angeles, California and received his B.S. degree in chemistry from UCLA in 1991. He earned his Ph.D. degree in chemistry with Dennis A. Dougherty at Caltech in 1997, and he performed postdoctoral research with Thomas R. Cech at the University of Colorado at Boulder. In 2000, he joined the Department of Chemistry at the University of Illinois at Urbana-Champaign, where he is currently Associate Professor of Chemistry. His research focuses on fundamental and applied studies of nucleic acids. This includes investigations of DNA as a catalyst, conformational constraint, and sensor as well as studies of RNA folding and catalysis. |
Introduction
Although DNA is identified most closely with its role in genetic information storage, considerations of its biophysical properties suggest application for other purposes. A wide range of experiments over the past two decades has explored the use of rigid double-stranded DNA (dsDNA) elements for “DNA nanotechnology”.1 For example, dsDNA has been used as a key component to build two- or three-dimensional objects that may be regular or irregular;2,3 to construct one- or two-dimensional lattices upon which other molecules assemble;4,5 and to devise molecular “machines” that undergo a variety of motions.6,7 However, none of these experiments exploit the rigidity of dsDNA to control the conformations of other discrete macromolecules. Recently, two research groups have reported the use of covalently attached dsDNA as a conformational constraint, using either proteins8–10 or RNA11,12 as the macromolecular target whose structure is controlled. These studies have validated the use of double-stranded DNA to control macromolecular structure and function. The results suggest new directions for the development of molecular tools that may be useful in biochemistry and biotechnology.Overview of DNA constraints for control of macromolecular structure
To illustrate the general DNA constraint approach, Fig. 1 includes two depictions that together apply to all of the experiments reported to date.8–12Fig. 1A shows a strategy in which a single DNA strand is connected covalently via both its 5′- and 3′-termini to a macromolecular target whose structure—and potentially function—will be controlled. Because single-stranded DNA (ssDNA) is rather flexible,13 the attached DNA is not anticipated to constrain the macromolecule's conformation, provided that the ssDNA is long enough to span the two attachment sites. However, upon addition of a separate DNA oligonucleotide that is complementary to a sufficient portion of the ssDNA, a rigid double-stranded DNA constraint is formed. If this duplex constraint is incompatible with the native macromolecular structure, then the impact of the constraint on folding and function should be detectable. Alternatively, Fig. 1B shows an approach in which two DNA strands are each connected via only their 5′-termini to a single macromolecular target (i.e., one connection to the macromolecule per DNA strand). If the two attached DNA strands are complementary to each other, then dsDNA formation can compete with adoption of the native macromolecular structure, thereby destabilizing that structure. This dsDNA formation will be destabilizing only if the dsDNA constraint is incompatible with the folded state of the macromolecule but not the unfolded state. For both approaches of Fig. 1, the energetic cost of disrupting the DNA duplex must be paid to gain the favorable folding energy of the macromolecule.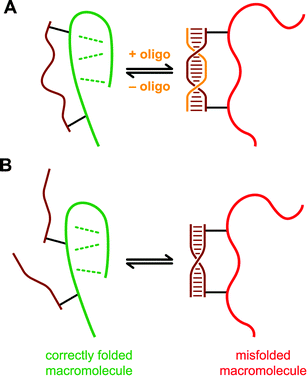 | ||
| Fig. 1 Two general strategies for using double-stranded DNA (dsDNA) to control macromolecular structure and function. (A) One DNA strand is connected covalently via both of its termini to the macromolecule. When a sufficiently complementary DNA oligonucleotide is added, formation of dsDNA alters the structure and function of the macromolecule. (B) Two complementary DNA strands are each connected by one terminus to different sites on the macromolecule. When the two DNA strands hybridize to each other and form dsDNA, the structure and function of the macromolecule are affected. In both illustrations, the dsDNA is compatible with the misfolded state of the macromolecule (right) but not the folded state (left). Therefore, the energy of dsDNA formation is lost when the macromolecule folds, and this folding is therefore destabilized. The DNA strands may not need to separate entirely to accommodate folding of the macromolecule, but such separation is shown here as a limiting case. | ||
DNA as a conformational constraint on protein structure and function
Zocchi and coworkers have recently reported three studies that focus on DNA as a protein-folding constraint.8–10 All of these experiments use the approach of Fig. 1A; the strategy of Fig. 1B has not yet been described for proteins. In the first study,8 dsDNA control was imposed on the structure of E. coli maltose-binding protein (MBP). MBP was chosen as the target protein because of its large, 10 Å conformational change upon binding of maltose—or a related ligand, maltotriose—in the cleft between the two lobes of the protein. Using one of two particular synthetic strategies, a 60 nucleotide ssDNA was attached via both of its termini to MBP. The first synthetic strategy used a single-cysteine mutant of MBP that also had an N-terminal his6 tag; the ssDNA was attached to the cysteine at one end via disulfide formation and at the other end to the his6 tag via metal complex formation. Alternatively, the second synthetic strategy used a double-cysteine mutant of MBP; each terminus of the ssDNA was attached to one of the cysteines via a heterobifunctional linker that allowed a terminal amino group on the DNA to be conjugated with a cysteine thiol. With suitably derivatized MBP available via either synthetic strategy, the ssDNA was converted to a suitable length of dsDNA by addition of a free DNA oligonucleotide complementary to a desired portion of the attached ssDNA. This reduced the maltotriose binding affinity of MBP, as assayed by tryptophan fluorescence determination of the binding constant (Kd). The magnitude of the effect on Kd was rather small. For example, in the most complete data set the Kd changed from ∼5.3 µM to ∼3.4 µM, which corresponds to a binding free energy difference (ΔΔG°) of ∼0.3 kcal mol−1.In the second study,9 a similar approach was applied to the enzyme guanylate kinase (GK), using the double-cysteine synthetic strategy. By using an enzyme rather than a binding protein as the target, the effect of a dsDNA constraint on protein function (rather than merely structure) could be determined (Fig. 2A). Specifically, luciferase chemoluminescence—which monitors the consumption of ATP concomitant with phosphorylation of the substrate GMP—was used to assess the deleterious effect of a DNA constraint upon catalysis by the protein enzyme. After establishment of the dsDNA constraint by addition of complementary DNA, a 4-fold decrease in “effective GK concentration” was observed. Fitting of the experimental data indicated that this corresponds to a 10-fold increase in the Km for substrate GMP (ΔΔG° ≈ 1.4 kcal mol−1) upon establishment of the dsDNA constraint. In both this study and the first report, the magnitude of the DNA constraint effect appears to be limited by the incomplete purity of the ssDNA-derivatized sample (estimated 50–70% purity), suggesting that improved synthetic methods for protein–DNA conjugation would be useful.
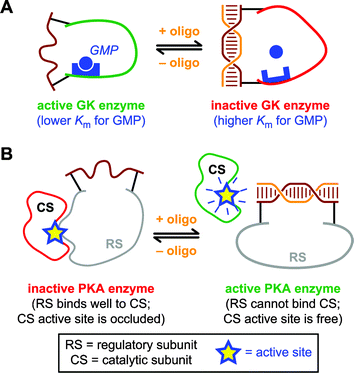 | ||
| Fig. 2 Schematic depictions of two studies in which protein structure and function were controlled using dsDNA via the approach of Fig. 1A. (A) Control of guanylate kinase (GK) activity.9 Enzyme activity is decreased several-fold when dsDNA is formed, likely because the binding site for substrate GMP becomes distorted. (B) Control of protein kinase A activity.10 Formation of dsDNA reduces the binding affinity of the regulatory subunit (RS) for the catalytic subunit (CS), similar to the allosteric effect of the natural activator cAMP. Therefore, formation of dsDNA increases the enzyme activity. | ||
In the third study,10 the enzyme protein kinase A (PKA) was linked with ssDNA using the double-cysteine synthetic strategy. In nature, PKA is allosterically controlled by binding of cyclic AMP (cAMP) to the regulatory subunit, which then dissociates from the catalytic subunit. The authors sought to mimic this cAMP-dependent allosteric control using their artificial DNA constraint (Fig. 2B). The results showed that kinase activity of the ssDNA–PKA chimera was stimulated either by cAMP itself or by the complementary DNA oligonucleotide. The efficiencies were comparable in terms of degree of stimulation when the modest (40–70%) purity of the chimera was taken into account. It should be noted that in this third study formation of the DNA duplex led to an increase in function (as designed), whereas in the first two efforts formation of dsDNA led to a decrease in function.
In all three of these studies in which a DNA constraint was attached to a protein, the findings were interpreted in terms of a physical model in which the dsDNA places mechanical tension upon the protein. This DNA constraint affects the structure of the protein, as assayed by Kd in the case of MBP. For GK and PKA, this effect on structure translates into an effect on catalytic activity, as reflected in the increase in Km for the substrate (for GK) or the upregulation of enzyme activity (for PKA).
DNA as a conformational constraint upon RNA structure
Miduturu and Silverman have published two reports on the use of dsDNA to control RNA folding.11,12 Both of these efforts used the general approach of Fig. 1B; related experiments that apply the alternative strategy of Fig. 1A are in progress (R. Morales and S. K. Silverman, unpublished results). In the first reported effort,11 two RNA strands were attached to the P4–P6 domain of the Tetrahymena group I intron RNA. P4–P6 is a 160 nucleotide independently folding RNA domain that is often used as a test system for exploring RNA structure and function.14–17 The RNA–DNA attachments were performed by reductive amination, using a 5′-tethered-aldehyde DNA and 2′-amino-RNA18 along with appropriate RNA–RNA ligation reactions. By choosing two DNA attachment sites on the P4–P6 RNA that were separated by ∼56 Å, which cannot be spanned by a short 10 bp dsDNA (∼34 Å), the RNA structure was expected to be destabilized because the DNA duplex must distort for the native RNA folding to occur (depicted schematically in Fig. 1B). Experimentally, substantial energetic destabilization of the RNA folding was indeed observed. The evidence was that the DNA-constrained P4–P6 RNA required a much higher Mg2+ concentration to fold according to nondenaturing polyacrylamide gel electrophoresis (native PAGE; Fig. 3). From the magnitude of the rightward shift in Mg2+ dependence, a ΔΔG° of ≥6 kcal mol−1 was estimated. Although this type of experiment does not reveal directly what physical distortion of the duplex DNA is necessary to achieve RNA folding, the magnitude of ΔΔG° is consistent with fraying of at least several DNA base pairs. This would allow sufficient “slack” to develop in the resulting portion of ssDNA, such that the RNA can subsequently fold properly. In contrast to these results with a 10 bp dsDNA constraint, when a 20 bp DNA duplex was placed across the same two RNA attachment sites, no destabilization was expected because the DNA was anticipated to be compatible with both the unfolded and folded RNA conformations; therefore, the free energy of DNA duplex formation should not affect the RNA folding equilibrium. Indeed, very little effect on RNA folding was observed with the 20 bp dsDNA.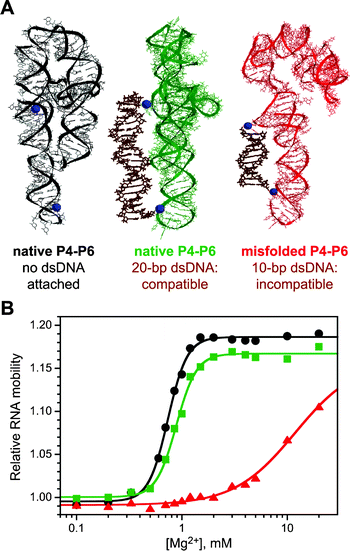 | ||
| Fig. 3 Control of P4–P6 RNA structure using dsDNA via the approach of Fig. 1B.11 (A) Molecular models of P4–P6. On the left is shown the native RNA conformation taken from the X-ray crystal structure15 with no DNA attached (for reference, the small spheres mark the sites of DNA attachment in the other two structures). In the middle is shown the native RNA conformation with a structurally compatible 20 bp dsDNA attached; the RNA conformation is essentially identical to the left structure but rotated ∼90° about a vertical axis so the DNA attachment sites can be seen clearly. On the right is shown a misfolded RNA conformation—one of many potential misfolded structures generated by modeling—with an incompatible 10 bp dsDNA attached. (B) Relative P4–P6 RNA mobility data from native PAGE show the rightward shift in Mg2+ dependence when two DNA strands that can form an incompatible 10 bp duplex are attached on appropriate RNA nucleotides. Circles, no DNA; squares, structurally compatible 20 bp dsDNA; triangles, incompatible 10 bp dsDNA (same left-to-right order as molecular structures in panel A). This rightward shift corresponds to a free energy destabilization (ΔΔG°) for RNA folding of ≥6 kcal mol−1. | ||
A large number of control experiments were performed to verify that the destabilization of RNA folding was due to the designed 10 bp dsDNA constraint. In particular, when the two attached DNA strands were not complementary, or when only one DNA strand was attached, essentially no effect on the Mg2+ dependence of RNA folding was observed. The integrity of RNA tertiary structure was also monitored by dimethyl sulfate (DMS) probing rather than native PAGE. The Mg2+ dependencies of RNA folding as determined by either technique were very similar, providing additional evidence that the DNA constraint itself (and not an artifact of any particular experimental method) was responsible for the RNA destabilization.
In the second study on DNA-constrained RNA folding, the structural effects of a DNA constraint were modulated in several ways, using added oligonucleotides, deoxyribonuclease, restriction enzymes, or reducing agents to disrupt the constraint.12 In the case of added oligonucleotides, fluorescence of a covalently attached pyrene chromophore19 was used to report on the reversible RNA folding–unfolding process as the DNA constraint was modulated. Perhaps the most intriguing experiment used a DNA constraint for which one of the two DNA strands was largely coincident with the known sequence of an aptamer for the porphyrin hemin20,21 (Fig. 4A). Addition of hemin to the DNA-constrained RNA released the constraint effect (Fig. 4B), as expected when the ligand–aptamer interaction disrupts the dsDNA constraint. This system is formally equivalent to a sensor because the presence or absence of hemin controls the structure of the RNA. A key advantage of the system is its modularity: in principle, a different aptamer sequence for another ligand could be used as part of the DNA constraint with the same RNA molecule. Of course, native PAGE is not a very practical signal readout for a realistic sensor, and future experiments in this area will focus on other signal readouts such as fluorescence or catalytic activity. The strategy depicted in Fig. 4 is related to “structure-switching signaling aptamers”,22 which similarly have an aptamer sequence that participates in duplex formation until the ligand is introduced. However, in this other approach to sensor systems, the duplex does not function to constrain macromolecular conformation as depicted in Fig. 4.
![Design and operation of a rudimentary sensor based on a dsDNA constraint and using RNA structure as the readout.12 (A) Design of the sensor, in which part of the dsDNA constraint is also an aptamer (bold line) for the hemin ligand that will be sensed. The nucleotide marked with an arrow is the site of a point mutation that is known to inactivate the aptamer for hemin binding (used here for control experiments). (B) Operation of the sensor, as shown by Mg2+ dependence of relative RNA mobility on native PAGE. The filled symbols show the dependence of relative RNA mobility on [Mg2+] in the presence of hemin when the aptamer is either the correct sequence (filled circles) or the mutant sequence (filled triangles). The open symbols show the results of the same experiment when performed in the absence of hemin. As expected, the RNA folds at relatively low [Mg2+] only when both hemin is present and the aptamer has the correct sequence. At an intermediate [Mg2+] such as 2 mM (vertical dashed line), the folding of the RNA reports on the presence or absence of hemin (filled vs. open circles).](/image/article/2007/MB/b614116a/b614116a-f4.gif) | ||
| Fig. 4 Design and operation of a rudimentary sensor based on a dsDNA constraint and using RNA structure as the readout.12 (A) Design of the sensor, in which part of the dsDNA constraint is also an aptamer (bold line) for the hemin ligand that will be sensed. The nucleotide marked with an arrow is the site of a point mutation that is known to inactivate the aptamer for hemin binding (used here for control experiments). (B) Operation of the sensor, as shown by Mg2+ dependence of relative RNA mobility on native PAGE. The filled symbols show the dependence of relative RNA mobility on [Mg2+] in the presence of hemin when the aptamer is either the correct sequence (filled circles) or the mutant sequence (filled triangles). The open symbols show the results of the same experiment when performed in the absence of hemin. As expected, the RNA folds at relatively low [Mg2+] only when both hemin is present and the aptamer has the correct sequence. At an intermediate [Mg2+] such as 2 mM (vertical dashed line), the folding of the RNA reports on the presence or absence of hemin (filled vs. open circles). | ||
The experiments on dsDNA control of RNA folding11,12 were primarily interpreted in terms of the simple geometrical (in)compatibility of the DNA duplex with the folded and unfolded RNA conformations (Fig. 1B). However, it is also reasonable to interpret the effects in terms of mechanical tension, as was done for the protein studies.8–10 In this view, the incompatible 10 bp dsDNA exerts tension on the folded P4–P6 RNA, which is relieved when the RNA unfolds. Equivalently, considering the RNA folding process in the forward direction, folding of P4–P6 must introduce tension into the RNA when the 10 bp dsDNA (but not 20 bp dsDNA) is present. The presence of covalently attached dsDNA that is incompatible with RNA tertiary structure may be similar to introducing mechanical tension onto RNA by other physical means.23
Stabilization (rather than destabilization) of macromolecular structure by dsDNA?
In contrast to destablizing macromolecular structure with dsDNA (Fig. 1), it should be possible instead to stabilize a macromolecule by judicious attachment of DNA strands in such a way that only the folded RNA state is compatible (Fig. 5). Indeed, such a result has recently been achieved for P4–P6 RNA folding, in which ∼2 kcal mol−1 stabilization was imparted to P4–P6 by strategic attachment of dsDNA.24 Such stabilization may be useful in biochemical studies (see below).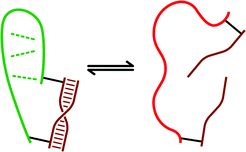 | ||
| Fig. 5 Strategy for selective stabilization of macromolecular conformation by dsDNA (compare with Fig. 1B). Here, the attached DNA duplex is compatible only with the folded state (left) and not the misfolded state (right) of the macromolecule. Therefore, the favorable energy of dsDNA formation adds to the macromolecule's folding energy. | ||
Potential biochemical and biotechnological applications of double-stranded DNA constraints
At least four potential applications of double-stranded DNA constraints await detailed exploration. First, dsDNA should be useful as a destabilizing influence to enforce a particular “misfolded” starting point for a folding experiment on a macromolecular folding landscape. In the case of RNA, misfolded states are known to serve as kinetic traps during folding,25 but it is a challenge to devise stable misfolded states with which to initiate a folding process.17 By strategically attaching dsDNA onto a larger macromolecule such as RNA, particular misfolded states should be created.Second, the ability to stabilize macromolecular folding by judicious attachment of DNA strands (Fig. 5) should enable dissection of individual energetic contributions to tertiary structure. This approach may be most useful for RNA, which generally has a hierarchical folding process.26 We envision that dsDNA can be attached to RNA in such a way as to replace a known tertiary contact such as a tetraloop–receptor interaction, which is very common in large folded RNAs.15,27 Then, it can be determined experimentally if the resulting DNA-stabilized RNA requires the initial tertiary contact, or if this tertiary contact is now dispensable for proper structure. Such experiments will relate directly to the roles and cooperativity of individual tertiary contacts in macromolecular structure.
Third, interpreting a dsDNA constraint as providing mechanical tension on another macromolecule suggests the application of covalently attached dsDNA for studying mechanotransduction of biochemical signals.28 For example, judiciously attached DNA duplexes may be able to mimic the effects of purely mechanical stress on proteins. The research by Choi and Zocchi with PKA already suggests that this is the case, inasmuch as ligand binding induces mechanical stress.10 It should be interesting to see if this general approach can be applied to mechanosensitive proteins that are naturally activated by mechanical means.
Finally, the combination of dsDNA constraints with generation of a molecular signal implies that dsDNA could be integrated into novel types of sensors. Much recent interest has focused on applications of “functional nucleic acids” as sensors.29 The successful integration of dsDNA constraints with ligand–aptamer interactions or oligonucleotide hybridization12 indicates that this overall approach may find utility for signal generation, particularly if more practical signal readout approaches such as fluorescence or catalysis can be developed. If issues of intracellular delivery and stability can be addressed, as is already being pursued for other applications of nucleic acids (e.g., antisense),30 it should additionally be possible to use such DNA-based sensors in vivo rather than solely in vitro. For example, a messenger RNA of interest could bind competitively to a dsDNA constraint, such that the catalytic activity of the macromolecule attached to the DNA is upregulated. If this catalysis is linked to a detectable event such as generation of a colored product or signal, then the presence of the mRNA will be reported.
Acknowledgements
Research in the Silverman lab is supported by the National Institutes of Health and by The David and Lucile Packard Foundation. I am grateful to Chandrasekhar V. Miduturu for initiation of our DNA constraint experiments. I thank Chandra Miduturu, J. P. Gerdt, Raymond Morales, Claudia Höbartner, and other members of the Silverman lab for many helpful discussions about DNA constraints.References
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef.
- W. M. Shih, J. D. Quispe and G. F. Joyce, Nature, 2004, 427, 618–621 CrossRef CAS.
- P. W. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- M. G. Warner and J. E. Hutchison, Nat. Mater., 2003, 2, 272–277 CrossRef CAS.
- H. Yan, S. H. Park, G. Finkelstein, J. H. Reif and T. H. LaBean, Science, 2003, 301, 1882–1884 CrossRef CAS.
- W. B. Sherman and N. C. Seeman, Nano Lett., 2004, 4, 1203–1207 CrossRef CAS.
- L. Jaeger and A. Chworos, Curr. Opin. Struct. Biol., 2006, 16, 531–543 CrossRef CAS.
- B. Choi, G. Zocchi, S. Canale, Y. Wu, S. Chan and L. J. Perry, Phys. Rev. Lett., 2005, 94, 038103 CrossRef.
- B. Choi, G. Zocchi, Y. Wu, S. Chan and L. J. Perry, Phys. Rev. Lett., 2005, 95, 078102 CrossRef.
- B. Choi and G. Zocchi, J. Am. Chem. Soc., 2006, 128, 8541–8548 CrossRef CAS.
- C. V. Miduturu and S. K. Silverman, J. Am. Chem. Soc., 2005, 127, 10144–10145 CrossRef CAS.
- C. V. Miduturu and S. K. Silverman, Angew. Chem., Int. Ed., 2006, 45, 1918–1921 CrossRef CAS.
- M. C. Murphy, I. Rasnik, W. Cheng, T. M. Lohman and T. Ha, Biophys. J., 2004, 86, 2530–2537 CrossRef CAS.
- F. L. Murphy and T. R. Cech, Biochemistry, 1993, 32, 5291–5300 CrossRef CAS.
- J. H. Cate, A. R. Gooding, E. Podell, K. Zhou, B. L. Golden, C. E. Kundrot, T. R. Cech and J. A. Doudna, Science, 1996, 273, 1678–1685 CrossRef CAS.
- J. P. Schwans, C. N. Cortez, J. M. Olvera and J. A. Piccirilli, J. Am. Chem. Soc., 2003, 125, 10012–10018 CrossRef CAS.
- C. Höbartner and S. K. Silverman, Angew. Chem., Int. Ed., 2005, 44, 7305–7309 CrossRef CAS.
- C. V. Miduturu and S. K. Silverman, J. Org. Chem., 2006, 71, 5774–5777 CrossRef CAS.
- M. K. Smalley and S. K. Silverman, Nucleic Acids Res., 2006, 34, 152–166 CrossRef CAS.
- Y. Li, C. R. Geyer and D. Sen, Biochemistry, 1996, 35, 6911–6922 CrossRef CAS.
- P. Travascio, Y. Li and D. Sen, Chem. Biol., 1998, 5, 505–517 CrossRef.
- R. Nutiu and Y. Li, J. Am. Chem. Soc., 2003, 125, 4771–4778 CrossRef.
- J. Liphardt, B. Onoa, S. B. Smith, I. J. Tinoco and C. Bustamante, Science, 2001, 292, 733–737 CrossRef CAS.
- Joseph P. Gerdt, Chandrasekhar V. Miduturu, Claudia Höbartner and Scott K. Silverman, manuscript in preparation.
- D. K. Treiber and J. R. Williamson, Curr. Opin. Struct. Biol., 2001, 11, 309–314 CrossRef CAS.
- I. Tinoco, Jr. and C. Bustamante, J. Mol. Biol., 1999, 293, 271–281 CrossRef CAS.
- M. Costa and F. Michel, EMBO J., 1995, 14, 1276–1285 CAS.
- V. Vogel, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 459–488 CrossRef CAS.
- N. K. Navani and Y. Li, Curr. Opin. Chem. Biol., 2006, 10, 272–281 CrossRef CAS.
- A. Tafech, T. Bassett, D. Sparanese and C. H. Lee, Curr. Med. Chem., 2006, 13, 863–881 CrossRef CAS.
Footnote |
| † The HTML version of this article has been enhanced with colour images. |
| This journal is © The Royal Society of Chemistry 2007 |