Controlling influenza virus replication by inhibiting its proton channel
Lawrence H.
Pinto
*a and
Robert A.
Lamb
b
aDepartment of Neurobiology and Physiology, Hogan Hall, 2205 Tech Drive, Northwestern University, Evanston, IL 60208-3500, USA. E-mail: larry-pinto@northwestern.edu; Fax: +1 (847)-491-5211; Tel: +1 (847) 491-7915
bDepartment of Biochemistry, Molecular Biology and Cell Biology and Howard Hughes Medical Institute, Northwestern University, Evanston, IL 60208-3500, USA
First published on 22nd November 2006
Abstract
Influenza A and viruses encode minimalistic proton-selective ion channels known as A/M2 and BM2, respectively. The A/M2 channel is the target of the antiviral drug, amantadine. The structural and mechanistic aspects of proton conductance of the viral ion channels are described and the review makes a case for the development of more effective antivirals.
 Lawrence H. Pinto | Lawrence H. Pinto, PhD is Professor of Neurobiology & Physiology at Northwestern University, Evanston |
 Robert A. Lamb | Robert A. Lamb, PhD, ScD is Professor of Biochemistry, Molecular Biology and Cell Biology at Northwestern University, Evanston, IL and an Investigator of the Howard Hughes Medical Institute. Lamb and Pinto have been collaborating to study influenza ion channels since 1990. |
Influenza A virus and influenza B virus are two related but distinct, enveloped, negative stranded RNA viruses that cause epidemic infections. The virus particles (virions) of each virus type contain a small integral membrane protein (A/M2 and BM2, respectively) that functions as a proton channel and is essential to viral replication. The A/M2 protein of influenza A virus is the target for the action of the antiviral drug amantadine, and thus this protein is important because it is a therapeutic target.1 Unfortunately, however, this drug does not inhibit replication of either influenza B virus or certain subtypes of influenza A virus. These proton channels are also of interest because they are among the smallest bona fideion channel proteins (with the properties of ion selectivity and activation), and they succeed in accomplishing the same function with only a meagre similarity in primary amino acid sequence. This similarity is contained in a single turn of the transmembrane helix that appears to impart the channels with their key proton transport properties. This review begins by discussing the roles of the ion channels in viral replication and summarizing their basic biochemistry and function. A mechanism for inhibition by amantadine is then proposed. This mechanism, together with knowledge of properties of amantadine-resistant ion channels, has lead to suggestions for inhibiting the amantadine-resistant ion channels.
The proton channels of both influenza A and B viruses must function in order for viral replication to occur (Fig. 1).2,3 Both viruses enter the infected cell by endocytosis, and when the virion is contained in the acidic environment of the lumen of the endosome, the interior of the virus particle becomes acidified. Acidification of the virion interior brings about the protein–protein dissociation that is required for proper uncoating of the virion, such that the viral genetic material can replicate.4,5,6 The proton channels mediate this acidification function.
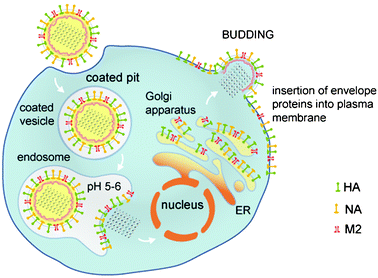 | ||
| Fig. 1 Life cycle of influenza virus showing the steps in which the M2 ion channel functions. The life cycle and role of the M2 protein are similar for both strains of influenza virus. The M2 proton channels function while the virion is contained in the endosome to permit subsequent uncoating. For certain subtypes of influenza A virus the M2 protein also shunts the pH gradient of the trans-Golgi apparatus to prevent premature conformational change in hemaglutinin. | ||
Basic biochemical properties of the proteins
Both A/M2 and BM2 proteins are homotetrameric, type III integral membrane proteins containing a small N-terminal ectodomain, a single transmembrane domain, and a C-terminal cytoplasmic tail. The transmembrane domain acts as both a signal sequence and a membrane anchor during protein synthesis. The predicted membrane-spanning domains of A/M2 and BM2 are 20 amino acids long and the N-terminal domain of the BM2 protein (7 residues) is shorter than that of the A/M2 protein (23 residues). Long cytoplasmic C-terminal domains characterize both the A/M2 protein (53 residues) and the BM2 protein (82 residues). The only homology between the amino acid sequences of these two proteins is found in the HXXXW motif of the inner membrane-spanning residues; this motif proves to be critical to the ion channel activity (see below). Both A/M2 and BM2 behave as homotetramers in cross-linking and sedimentation experiments,7,8,4,9 and the active oligomeric state of the A/M2 protein was demonstrated to be a tetramer.10 The A/M2 protein is post-translationally modified by palmitoylation and phosphorylation but these modifications do not seem to be important for ion channel function.11 Thus, the flux of protons across the membrane must occur within the pore formed by the four identical subunits of the transmembrane domain of the protein. The mechanism by which protons traverse the pore is the subject of active investigation.Functional mechanism for proton transport
Both ion channels are very selective for protons.12,13,14,15,16 The selectivity of A/M2 depends on a histidine residue in the transmembrane domain. Ion selectivity measurements have been made using in vitro expression systems12,14,15,17,18 and by reconstitution of ion channel activity from recombinant protein in bilayers19,20 or liposomes.13 The high proton selectivity of the channel is lost when transmembrane domain His37 is replaced with glycine, alanine, glutamic acid, serine or threonine,21,22 making the mutant channel capable of transporting Na+ and K+ as well. The ion selectivity of histidine substitution mutant proteins is partially restored by adding imidazole buffer to the solution bathing the expressing cell.21 The δ-nitrogen of imidazole has been shown to be protonated at low pH; this finding was obtained in solid-state NMR experiments using the transmembrane peptide from the M2.23 Thus, the imidazole side chain of histidine plays an essential role in the specificity for proton transport. Furthermore, it is possible that nitrogen atoms of the imidazole side chain interact with the protons that pass through the channel pore, as the pKa of histidine is ca. pH 7.The mechanism for the transport of protons, from outside the virion to inside, through the aqueous pore of the channel, has not been established with certainty, but two observations on the A/M2 channel are informative. First, the specific activity (single channel conductance) of the wild-type (wt) A/M2 ion channel is very low (it transports roughly 105 protons per tetramer per second at pH 5.7, the pH found in endosomes).13,50 Second, the kinetic isotope effect ,50 measured when deuterium replaces hydrogen, has a value of 1.8–2.5. This result shows that conductance decreases by an amount greater than the ratio of the diffusion coefficients of the two isotopes . These observations suggest that bulk transport of hydronium ions is not responsible for proton transport.50 Two other mechanisms have been suggested for proton transport. First, imidazole might serve as a “relay” molecule, binding protons presented from one end of the channel and releasing them to the other end by dissociation; this mechanism might be assisted by tautomerization of the imidazole.25 Second, short-lived proton ‘wires’ might open to allow shuttling of protons from one water molecule to another in the pore, without the water molecules themselves moving.50 Energy minimization simulations support the former model26 and molecular dynamics simulations support the latter model.27 Although there is little similarity between the amino acid sequences of the transmembrane domains of the A/M2 and BM2 channels (see below), it is presumed that the BM2 channel functions by a similar mechanism to the A/M2 channel because replacement of histidine in the transmembrane domain renders the channel nonfunctional. Thus, the exact mechanism for transport of protons with high selectivity is not known.
Mechanism for opening of the A/M2 ion channel
The A/M2 channel only conducts protons when the pH of the medium bathing the N-terminal ectodomain, pHout, is lowered below ca. pH 7. This ability to open and close is dependent on the action of a single transmembrane domain residue, Trp41. Two observations suggest that the channel is closed when pHout exceeds pH 7.5 and is opened when pHout is lower than pH 6.5. First, oocytes that express the channel become acidified rapidly when they are bathed in solutions of low pH but upon restoration of pHout to its normal value their internal pH recovers only very slowly. This restoration of pH occurs much more slowly than the re-alkalinization of cells treated with the protonophore FCCP,14,50 suggesting efflux of protons from M2 expressing cells is impaired by elevated pHout. Second, cells expressing A/M2 protein that are injected with acid (e.g., 1 M HCl) while pHout is above pH 7.5 do not experience an efflux of protons, but when pHout is low, efflux of protons can be brought about by applying a positive voltage to the inside of the cell.12,14,17,16 Thus, high pHout closes the channel and low pHout opens (activates) the channel.Several lines of evidence point to Trp41 (Fig. 2) as the key residue in opening and closing the channel pore.28 (1) For the wt ion channel, outward currents are not observed when pHout is high, regardless of the means taken to establish an outward electrochemical gradient for protons. Unlike the case for the wt ion channel, it is possible to observe outward proton currents under these conditions for mutant ion channel proteins in which Trp41 is replaced with amino acids having a small side-chain. (2) It is possible to improve the closing of a mutant ion channel in which Trp41 is replaced with Cys by placing a functional group resembling the Trp side chain on the Cys sulfur atom. (3) Cu(II) injected intracellularly is able to coordinate with His37 in a mutant ion channel in which Trp is replaced by Ala, but is not able to coordinate with His37 when injected into cells expressing the wt channel. An explanation for these observations is that the bulky indole side chain of Trp41 interferes with the passage of protons when pHout is high.28 This interpretation is supported by oxidative disulfide cross-linking analysis showing that a structural rearrangement occurs in this region of the protein when pHout is altered.29 Furthermore, Raman resonance spectroscopy has shown that pH-dependent interactions occur between His37 and Trp41, perhaps between the protonated imidazole and the π-electrons of the indole.30 Thus, opening and closing of the channel depends on pHout and probably involves structural alterations that encompass Trp41. It is noteworthy that the key functional elements of the channel, His37 and Trp41, are contained in a single turn of the transmembrane helix of this very compact channel.
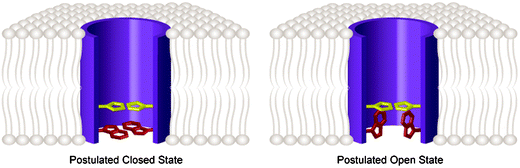 | ||
| Fig. 2 Model for proton transport and opening of the M2 ion channel showing only the TM domain from residues 24 to 44 (purple); the selectivity filter His37 (yellow), and the gate Trp41 (red) are highlighted. For clarity these residues are shown for only two of the four subunits; the residues facing the viewer and the subunit closest to the viewer are omitted. Left shows the scheme for the closed wt M2 channel. The channel is closed when pHout is high because His37 is not charged and Trp41 obstructs the pore near its cytoplasmic end. Right shows the wt M2 channel in the open state. With low pHout His37 is charged, allowing rotation of Trp41 to a conformation parallel to the pore's axis, permitting H+ to flow. In this state the size of the imidazole side chains from the four subunits obstructs the pore for ions such as Na+ and K+ but permits H+ presented from one side to be bound and subsequently released to the other side. | ||
Identification of the pore-lining residues of the A/M2 ion channel
Because protons must pass through the membrane via the pore of an ion channel, it is very important to identify the residues that line the pore and to obtain a rough idea of the secondary structure of the transmembrane domain. The former has been done for the A/M2 ion channel by combining cysteine scanning mutagenesis and water soluble sulfhydryl specific reagents that attach a large hydrophobic adduct to cysteine residues.31 If the conductance of a mutant channel with a cysteine residue in a particular location is diminished by the reagent, then it is concluded that the sulfhydryl moiety of the cysteine faces the aqueous pore at that location. The conductances of cysteine mutant proteins A30C and G34C were diminished when the reagent was applied to the medium bathing of the N-terminal ectodomain. The conductance of the cysteine mutant protein W41C was decreased when the reagent was injected into the cytoplasm but not when applied to the bathing medium. Moreover, the G34C mutant protein was not affected by cytoplasmic injection of the reagent. These results are consistent with His37 forming a barrier to large molecules and are also consistent with the proposed role for Trp41 as a gate capable of closing the channel. Cysteine scanning mutagenesis was also used together with oxidative disulfide cross linking29 to show that residues 27, 30, 34, 37 and 41 formed dimers most rapidly. Most interesting, however, was the finding that when oxidation was performed at low pHout (pH 5.2) the rate of cross linking for residues 40, 42 and 43, relative to the formation of cross linking for other residues, was much lower than when performed at neutral pHout (pH 7.4). This dependence of the relative rate of cross linking for residues near Trp41 suggests that a pH-dependent alteration of conformation occurs in this region of the molecule. The secondary structure of the transmembrane domain of the A/M2 channel has been studied with solid state NMR spectroscopy of the transmembrane peptide32,33,34,35 and the full length A/M2 ion channel protein.38,37 These studies concluded that the orientation of the helical bundle forming the transmembrane domain has a tilt angle of approximately 25°, relative to a normal to the membrane, and that transmembrane domain Trp of one subunit interacts with His of an adjacent subunit.33 Furthermore, hydrogen–deuterium exchange measurements showed that residues in the transmembrane helix exchange more rapidly than residues located in the cytoplasmic domain, consistent with the presence of an aqueous pore. Thus, the aqueous pore of the A/M2 ion channel is formed by Ala30, Gly34, His37 and Trp41, and these residues are found on a transmembrane helical bundle having a tilt angle of ca. 25 degrees.The cytoplasmic tail of the A/M2 ion channel
The cytoplasmic tail is the largest domain of both the A/M2 and BM2 proteins, and this domain has been found to be essential for the function of the A/M2 channel. Truncation of the A/M2 channel at residue 61 or shorter resulted in mutant ion channels with activity that could not be sustained.38 Solid state NMR experiments have provided the first direct structural information about the cytoplasmic domain for residues 45–62.36 These studies indicated that an amphipathic helix is found in this region, associated with the inner membrane leaflet. This amphipathic helix was found in the same region of the cytoplasmic tail that had been found to be essential for ion channel activity.38 Thus, even though ion channel activity has been demonstrated in lipid bilayers using only peptides that mimic the A/M2 transmembrane domain,39 the cytoplasmic tail certainly plays an essential role in the function of the wt ion channel protein.Summary of structure–function relationship of the A/M2 ion channel
The available biochemical and solid state NMR data indicate that the transmembrane domain of the A/M2 protein is comprised of a four helix bundle with a tilt of about 25°. These experiments are also consistent with the functional studies that showed that residues Val27, Ala30, Gly34, His37 and Trp41 line the aqueous pore. Furthermore, these results indicate that His37 forms a barrier to large molecules and that Trp41 functions as a gate that closes with high pHout. Functional studies indicate that the portion of the cytoplasmic domain nearest the transmembrane domain is important for normal ion channel function, and solid state NMR studies indicate that the structure of this domain brings it into close proximity to the inner membrane leaflet. The most remarkable aspect of this channel are the observations that much of its functionality is provided for by the His37 and Trp41 residues found in one turn of the transmembrane helical bundle.Inhibition of the A/M2 ion channel
This topic is important because antiviral drugs that inhibit the M2 ion channel also inhibit replication of the influenza A virus, and understanding the mechanism of inhibition would be helpful in developing better inhibitors. The antiviral drug amantadine and its derivative rimantadine inhibit the replication of the influenza A virus but not influenza B virus.40Amantadine inhibits the A/M2 ion channel,12,17,41 but not the BM2 channel,15 which is consistent with amantadine inhibiting influenza A virus but not influenza B virus replication. Several lines of evidence indicate that inhibition of viral replication by amantadine results from the inhibition of A/M2 proton channel activity. The first evidence comes from amantadine-resistant escape mutant viruses in which the site of the mutation was mapped and found to lie in the A/M2 transmembrane domain.42 When these escape mutation proteins were expressed in oocytes43,17 or mammalian cells12,18 their currents were found to be insensitive to amantadine. The second line of evidence comes from consideration of the virus life cycle. Viral uncoating requires acidification of the virus prior to fusion with the endosomal membrane44,45,24 and the M2 protein is capable of providing the needed acidification when the virion is contained in the endosome. The third line of evidence is that the viral genome encodes only three integral membrane proteins46 and only A/M2 is capable of proton transport. Thus, inhibition of the essential proton transport function of the A/M2 ion channel results in inhibition of replication of the virus.Five observations, taken together, suggest a mechanism for inhibition by amantadine (Fig. 3). (1) Two of the mutations that result in resistance to amantadine occur on residues that have been found by cysteine scanning mutagenesis to line the aqueous pore. Both of these mutations are to residues that are less hydrophobic (A30T and G34E) than the native residue.42 (2) Inhibition of the channel occurs more readily when the pH of the bathing medium is high,.41 (3) Amantadine only inhibits channel activity when it is applied to the medium bathing the N-terminal ectodomain, and not when applied to the C-terminal cytoplasmic tail; injection of as much as 1 mM amantadine into cells expressing the A/M2 channel does not inhibit the channel, whereas application of 10 µM to the solution bathing the ectodomain inhibits the channel fully.41 (4) Neutron diffraction studies of amantadine applied to the M2 transmembrane peptide show the compound to lie in the outer region of the membrane.47 (5) Amantadine inhibits with a 1 : 1 stoichiometry.41 These observations suggest that amantadine acts from the outside of the aqueous pore, that the hydrophobic adamantane group interacts with hydrophobic pore-lining residues (Ala30 and Gly34) and that perhaps the ammonium nitrogen of amantadine shares a hydrogen bond with a nitrogen atom of an unprotonated imidazole side chain of histidine.14 If such hydrogen bonding occurred it would interrupt the interactions formed by the ring of His and Trp residues from adjacent subunits.33 Thus, amantadine probably resides within the aqueous pore of the channel when it inhibits but probably does not inhibit by simply blocking the pore.
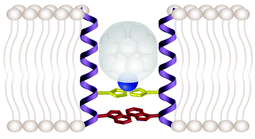 | ||
| Fig. 3 Postulated mechanism for inhibition of the A/M2 ion channel by amantadine. The hydrophobic adamantane moiety of amantadine (white) is shown associated with the hydrophobic side chains of the pore-lining residues of amantadine-sensitive an channel. The amide nitrogen of amantadine (Blue) and a nitrogen from the imidazole side chain of histidine are shown in proximity by hydrogen bonding. Perhaps this surrogate proton prevents the flow of free protons and inhibits channel activity. Amantadine-resistant A/M2 channels and the BM2 channel have in common the presence of one or more polar pore-lining residues. | ||
Why doesn't amantadine inhibit the BM2 ion channel?
It is useful to compare the amino acid sequences of the outer region of the transmembrane domain of these two ion channelsbecause amantadine inhibits the A/M2 channel by acting in the outer region of the pore (see above). The pore-lining residues of A/M2 have been identified in previous studies.48,31 It is reasonable to accept the conclusion that His19 and Trp23 of the BM2 protein are pore-lining residues because mutation of these residues causes alterations in BM2 function that are similar to those found by mutation of His37 and Trp41 in the A/M2 ion channel. Accepting these residues as pore-lining and assuming that the BM2 transmembrane domain (underlined in sequences below) forms a helical bundle, alignment of the sequences of the two channels shows that polar serine residues occur in the BM2 channel in locations that are predicted to be pore-lining:
The presence of polar residues would be expected to reduce the affinity of the hydrophobic adamantane moiety of the inhibitor. It is of interest to note that replacement of a hydrophobic residue with a polar residue occurs in an amantadine-resistant escape mutation of the A/M2 ion channel. The mutation M2-A30T is amantadine resistant and in fact, quite active.43 Thus, the reason that the BM2 ion channel is not inhibited by amantadine is probably because the BM2 channel pore region is lined with polar, and not hydrophobic, amino acids. This explanation shows that the inability of amantadine to inhibit the BM2 protein is understandable in terms of the primary amino acid sequence of its transmembrane domain.
Improving upon amantadine
Two classes of compounds are known to inhibit replication of influenza virus, amantadine-like compounds that inhibit the M2 ion channel and prevent viral uncoating and compounds that inhibit the neuraminidase enzyme, the activity of which is essential for viral budding.1 Mutations that permit the virus to escape from the effects of amantadine occur readily and these mutations do not tend to impart the virus with decreased fitness because the mutations do not detectably lower the activity of the ion channel.43 In contrast, mutations that permit the neuraminidase enzyme to escape from the effects of inhibitor compounds tend to have reduced fitness.49 The reason for this difference is probably that neuraminidase inhibitors were designed to interact with the active site of the enzyme and mutations that alter the active site enough to escape the effect of the inhibitor will inevitably affect the function of the enzyme. On the other hand, according to our model, amantadine inhibits the M2 ion channel by interacting with hydrophobic residues in the pore and sharing a hydrogen bond with the essential imidazole side chain of histidine. Amantadine-resistant escape mutations lessen these hydrophobic interactions. Several ways come to mind for skirting this problem. First, the highly conserved HXXXW motif might be exploited; if a compound could be created that interacts with the conserved His and Trp residues it might be possible to force reduced ion channel function upon escape mutants. Second, it might be possible to create a compound that interacts with hydrophilic pore-lining residues and use this compound for amantadine-resistant subtypes such as the H5N1 avian subtype that has proven lethal in a high percentage of infected persons. Although these suggestions sound promising based on our present knowledge of the pore of the ion channel, it is certain that detailed structural information will make it more likely that much better inhibitors can be found.Acknowledgements
We thank C. Niemiec for help with the illustrations. R.A.L. is an Investigator of the Howard Hughes Medical Institute. Research in the authors' laboratories is supported by research grants R01AI-31882, R01-AI57362 and R01-AI-71342 (LHP) and R37AI-20201 (RAL) from the National Institutes of Allergy and Infectious Diseases.References
- R. A. Lamb and R. M. Krug, “Orthomyxoviridae: the viruses and their replication.” in, Fields Virology, ed D. M. Knipe and P. M. Howley, Lippincott, Williams and Wilkins, Philadelphia, 2001, pp. 1487–1531 Search PubMed.
- D. Jackson, A. Cadman, T. Zurcher and W. S. Barclay, J. Virol., 2002, 76, 11744–11747 CrossRef CAS.
- M. Takeda, A. Pekosz, K. Shuck, L. H. Pinto and R. A. Lamb, J. Virol., 2002, 76, 1391–1399 CAS.
- R. G. Paterson, M. Takeda, Y. Ohigashi, L. H. Pinto and R. A. Lamb, Virology, 2003, 306, 7–17 CrossRef CAS.
- O. P. Zhirnov, Virology, 1990, 176, 274–279 CrossRef CAS.
- O. P. Zhirnov, Virology, 1992, 186, 324–330 CrossRef CAS.
- L. J. Holsinger and R. A. Lamb, Virology, 1991, 183, 32–43 CrossRef CAS.
- P. P. Panayotov and R. W. Schlesinger, Virology, 1992, 186, 352–355 CrossRef CAS.
- R. J. Sugrue and A. J. Hay, Virology, 1991, 180, 617–624 CrossRef CAS.
- T. Sakaguchi, G. P. Leser and R. A. Lamb, J. Cell Biol., 1996, 133, 733–747 CrossRef CAS.
- L. J. Holsinger, M. A. Shaughnessy, A. Micko, L. H. Pinto and R. A. Lamb, J. Virol., 1995, 69, 1219–1225 CAS.
- I. V. Chizhmakov, F. M. Geraghty, D. C. Ogden, A. Hayhurst, M. Antoniou and A. J. Hay, J. Physiol., 1996, 494, 329–336.
- T. I. Lin and C. Schroeder, J. Virol., 2001, 75, 3647–3656 CrossRef CAS.
- J. A. Mould, J. E. Drury, S. M. Frings, U. B. Kaupp, A. Pekosz, R. A. Lamb and L. H. Pinto, J. Biol. Chem., 2000, 275, 31038–31050 CrossRef CAS.
- J. A. Mould, R. G. Paterson, M. Takeda, Y. Ohigashi, P. Venkataraman, R. A. Lamb and L. H. Pinto, Dev. Cell, 2003, 5, 175–184 CrossRef CAS.
- K. Shimbo, D. L. Brassard, R. A. Lamb and L. H. Pinto, Biophys. J., 1996, 70, 1336–1346 Search PubMed.
- L. H. Pinto, L. J. Holsinger and R. A. Lamb, Cell, 1992, 69, 517–528 CrossRef CAS.
- C. Wang, R. A. Lamb and L. H. Pinto, Virology, 1994, 205, 133–140 CrossRef CAS.
- M. T. Tosteson, L. H. Pinto, L. J. Holsinger and R. A. Lamb, J. Memb. Biol., 1994, 142, 117–126 Search PubMed.
- V. Vijayvergiya, R. Wilson, A. Chorak, P. F. Gao, T. A. Cross and D. D. Busath, Biophys. J., 2004, 87, 1697–1704 CrossRef CAS.
- P. Venkataraman, R. A. Lamb and L. H. Pinto, J. Biol. Chem., 2005, 280, 21463–21472 CrossRef CAS.
- C. Wang, R. A. Lamb and L. H. Pinto, Biophys. J., 1995, 69, 1363–1371 CrossRef CAS.
- J. Hu, R. Fu, K. Nishimura, L. Zhang, H. X. Zhou, D. D. Busath, V. Vijayvergiya and T. A. Cross, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6865–6870 CrossRef CAS.
- K. Martin and A. Helenius, Cell, 1991, 67, 117–130 CrossRef CAS.
- L. H. Pinto, G. R. Dieckmann, C. S. Gandhi, M. A. Shaughnessy, C. G. Papworth, J. Braman, J. D. Lear, R. A. Lamb and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 11301–11306 CrossRef CAS.
- J. D. Lear, FEBS Lett., 2003, 552, 17–22 CrossRef CAS.
- A. M. Smondyrev and G. A. Voth, Biophys. J., 2002, 83, 1987–1996 CrossRef CAS.
- Y. Tang, F. Zaitseva, R. A. Lamb and L. H. Pinto, J. Biol. Chem., 2002, 277, 39880–39886 CrossRef CAS.
- C. M. Bauer, L. H. Pinto, T. A. Cross and R. A. Lamb, Virology, 1999, 254, 196–209 CrossRef CAS.
- A. Okada, T. Miura and H. Takeuchi, Biochemistry, 2001, 40, 6053–6060 CrossRef CAS.
- K. Shuck, R. A. Lamb and L. H. Pinto, J. Virol., 2000, 74, 7755–7761 CrossRef CAS.
- F. A. Kovacs, J. K. Denny, Z. Song, J. R. Quine and T. A. Cross, J. Mol. Biol., 2000, 295, 117–125 CrossRef CAS.
- K. Nishimura, S. Kim, L. Zhang and T. A. Cross, Biochemistry, 2002, 41, 13170–13177 CrossRef CAS.
- Z. Song, F. A. Kovacs, J. Wang, J. K. Denny, S. C. Shekar, J. R. Quine and T. A. Cross, Biophys. J., 2000, 79, 767–775 CrossRef CAS.
- J. Wang, S. Kim, F. Kovacs and T. A. Cross, Protein Sci., 2001, 10, 2241–2250 CrossRef CAS.
- C. Tian, P. F. Gao, L. H. Pinto, R. A. Lamb and T. A. Cross, Protein Sci., 2003, 12, 2597–2605 CrossRef CAS.
- C. Tian, K. Tobler, R. A. Lamb, L. H. Pinto and T. A. Cross, Biochemistry, 2002, 41, 11294–11300 CrossRef CAS.
- K. Tobler, M. L. Kelly, L. H. Pinto and R. A. Lamb, J. Virol., 1999, 73, 9695–9701 CAS.
- K. C. Duff and R. H. Ashley, Virology, 1992, 190, 485–489 CrossRef CAS.
- W. L. Davies, R. R. Grunert, R. F. Haff, J. W. McGahen, E. M. Neumayer, M. Paulshock, J. C. Watts, T. R. Wood, E. C. Herman and C. E. Hoffman, Science, 1964, 144, 862–863 CrossRef CAS.
- C. Wang, K. Takeuchi, L. H. Pinto and R. A. Lamb, J. Virol., 1993, 67, 5585–5594 CAS.
- A. J. Hay, A. J. Wolstenholme, J. J. Skehel and M. H. Smith, EMBO J., 1985, 4, 3021–3024 CAS.
- L. J. Holsinger, D. Nichani, L. H. Pinto and R. A. Lamb, J. Virol., 1994, 68, 1551–1563 CAS.
- M. Bui, G. Whittaker and A. Helenius, J. Virol., 1996, 70, 8391–8401 CAS.
- A. Helenius, Cell, 1992, 69, 577–578 CrossRef CAS.
- R. A. Lamb, S. L. Zebedee and C. D. Richardson, Cell, 1985, 40, 627–633 CrossRef CAS.
- K. C. Duff, P. J. Gilchrist, A. M. Saxena and J. P. Bradshaw, Virology, 1994, 202, 287–293 CrossRef CAS.
- C. S. Gandhi, K. Shuck, J. D. Lear, G. R. Dieckmann, W. F. DeGrado, R. A. Lamb and L. H. Pinto, J. Biol. Chem., 1999, 274, 5474–5482 CrossRef CAS.
- R. R. Regoes and S. Bonhoeffer, Science, 2006, 312, 389–391 CrossRef CAS.
- J. A. Mould, H.-C. Li, C. S. Dudlak, J. D. Lear, A. Pekosz, R. A. Lamb and L. H. Pinto, J. Biol. Chem., 2000, 275, 8592–8599 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |