Design of radiolabelled ligands for the imaging and treatment of cancer
Stephen J.
Mather
Centre for Cancer Imaging and Queen Mary School of Medicine and Dentistry, St Bartholomew's Hospital, London EC1A 7BE, UK. E-mail: Stephen.Mather@cancer.org.uk
First published on 14th November 2006
Abstract
The specialised medical field of Nuclear Medicine is concerned with the use of unsealed sources of radioactivity either to diagnose or treat a range of diseases. In this regard it can be distinguished from the field of Radiotherapy which uses sealed radioactive sources for treatment. The range of diseases in which Nuclear Medicine plays a role is wide and includes, among others, the fields of microbiology, endocrinology, neurology, oncology and cardiovascular medicine. However, cancer probably represents the most important and growing area of application for this modality. Nuclear Medicine employs radiopharmaceuticals. These are radiolabelled ligands that have the ability to interact with molecular targets that are relevant in the aetiology or treatment of cancer and in many respects Nuclear Medicine can be considered the archetype for the application of ‘Molecular Medicine’. An example of a Nuclear Medicine Positron Emission Tomography (PET) scan is shown in Fig. 2. There is great interest in developing new radioligands that allow us to image the expression of the ever increasing range of biological pathways being discovered in the post-genomic area. Designing effective radiopharmaceuticals, however, requires an understanding of a number of radiopharmaceutical sciences including aspects of chemistry, physics, cell and molecular biology, and physiology.
 Stephen Mather | Professor Stephen Mather has worked as a Radiopharmacist in either hospitals or universities for the past 30 years. His research has been funded for the past 25 years by Cancer Research UK (previously Imperial Cancer Research Fund). He is currently Head of Cancer Imaging in the Institute of Cancer, Barts and the London, Queen Mary School of Medicine and Dentistry in London, UK |
1. Introduction
The perfect radiopharmaceutical (if it existed) would, after intravenous administration, travel via the blood stream to the target cells where it would efficiently interact solely with the desired molecular pathway. Any radioligand that did not reach the desired target would be rapidly excreted from the body so that the only radioactivity remaining in the body would be that localised to the target site. The objective in radiopharmaceutical design is, therefore, to get as close as possible to this ideal situation. The radiopharmaceutical can be considered to have three components: (i) the targeting vector, (ii) the radionuclide and (iii) a means of linking the two together that may, or may not, include a pharmacokinetic modifier. An example is shown in Fig. 1.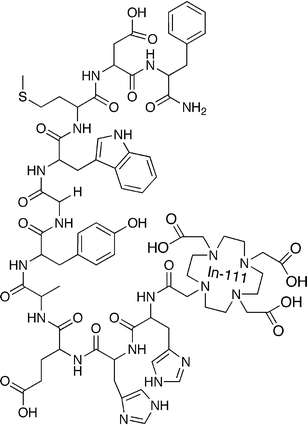 | ||
| Fig. 1 An example of the structure of a receptor-binding peptide-based radioligand. From top counter-clockwise, the first eight amino acid residues make up the sequence that binds to the gastrin receptor. The di-His linker reduces the kidney retention of the conjugate. The DOTA chelator binds the radionuclide, Indium-111 (shown not complexed by the DOTA), which allows imaging of the biodistribution of the conjugate by SPECT. | ||
2. Targets for radiopharmaceutical development
Any biological target that is present at increased (preferably) or decreased levels in cancer cells can be usefully pursued. Since there are hundreds of pathways that are implicated in malignancy, a more discriminating selection can be made as follows: ideally the target should be as specific as possible for the disease i.e. not present in normal tissues or those involved in other diseases such as inflammation and should be present as abundantly as possible on cancer cells. In selecting a target, consideration should also be given as to the clinical information that would be gained from a diagnostic or therapeutic procedure that interacts with this pathway. Thus the requirements for developing an imaging procedure that provides a sensitive and specific means for staging cancer on first presentation will be different to those required for assessing the response of a particular type of cancer to a particular therapeutic intervention.Targets of current interest in radiopharmaceutical development can be divided into two main (overlapping) categories: (i) cell surface markers or receptors that show significantly increased level of expression on malignant cells, and (ii) intracellular metabolic pathways that are either up-regulated in cancer or are implicated in the response to various types of treatment.
2.1 Cell surface binding ligands
The requirements for binding targets at the cell surface are less stringent than those required to pass through the cell membrane. Ligands currently being pursued are those based on monoclonal antibodies which bind to tumour-associated epitopes and neuropeptide hormones that bind to a range of neuropeptide receptors. Some examples of each type are given in Table 1. The earliest exploitation of radiolabelled monoclonal antibodies occurred a few years after their initial development at the beginning of the 1980's. It was shown that these relatively large proteins could specifically interact with their complementary epitopes in vivo but it also became apparent that they were far from ideal targeting vectors.1 Physical access to the tumour cells was limited by poor blood supply and a number of physical barriers because of their large size which also resulted in a slow rate of clearance from the blood stream.2 Normal organs such as the kidney and liver accumulated much larger amounts of radioactivity than most tumours and the xenogenetic origin of the antibodies (normally mice) resulted in their recognition as foreign proteins and production of neutralising antibodies by the recipient's immune system.3 In the intervening two decades, development has centred on the need to reduce the size and immunogenicity of these potentially useful molecules. The problem of immune recognition was solved by replacement of mouse framework regions in the protein, by those derived from human antibody sequences.4 This has resulted in the development of a large number of antibody-based drugs that are currently becoming approved for clinical use5 but these are still full-size proteins that are likely to be unsuitable as radiopharmaceuticals. The problem of size has been addressed by removing most of the superfluous framework regions of the immunoglobulin while retaining the epitope -binding domains6 or by screening libraries composed of molecules based on much smaller scaffolds. A number of different strategies have been employed.7 Attempts have also been made to increase the avidity of target binding (and stability of the protein) by oligomerisation of these small binding domains.8| Molecular target | Type of target | Ligand(s) | Reference |
|---|---|---|---|
| Somatostatin receptor | Neuropeptide receptor | Octreotide analogues | 10 |
| Gastrin releasing peptide receptor | Neuropeptide receptor | Bombesin analogues | 13 |
| CD-20 | Lymphoblastoid cell-surface receptor | Monoclonal antibodies against CD-20 | 14 |
| Carcinoembryonic antigen | Oncofoetal epitope | Monoclonal antibodies against CEA | 15 |
| Epithelial growth factor receptor | Growth factor receptor | EGF | 16 |
| Prostate specific membrane antigen | Tissue specific epitope | Monoclonal antibodies against PSMA | 17 |
| αvβ6 integrin | Angiogenesis associated integrin | Cyclic RGD peptides | 18 |
| Phosphatidylserine | Apoptosis | Annexin analogs | 19 |
To some extent this drive towards miniaturised antibodies has been driven by the success in tumour targeting of small radiolabelled peptide hormones, receptors for which are expressed on many tumours.9 The paradigm for this approach is the application of radiolabelled octreotide analogues for targeting somatostatin receptors which show increased levels of expression by a number of tumour types.10 These ligands lack many of the disadvantages of antibodies: they are small, typically 100 times smaller than IgG. This allows them to diffuse more easily into the tumour environment and also means that they clear more rapidly from the bloodstream and, because they are of human origin, they are not recognised by the patient's immune system. However, they do possess disadvantages of their own: many naturally occurring neuropeptide hormones are unstable in the bloodstream being rapidly degraded by serum proteases. This is a natural mechanism of limiting the duration of their normal pharmacological action. In addition, this pharmacological activity can lead to undesirable side effects even at very low doses. To improve the stability, the sequence of the natural peptide can be modified by insertion of unnatural amino acids or modification of peptide bonds, provided that this does not disrupt their interaction with the receptor.11 The pharmacological activity can be viewed from both negative and positive angles. The fate of most pharmacological agonists after binding to the receptor is that the receptor-ligand complex is internalised and packed in intracytoplasmic vesicles from which the receptor is re-cycled to the cell surface but in which the ligand is degraded.12 Depending on the radionuclide being used, this can result in it being trapped within the cell for the duration of its physical decay. This long retention time can greatly enhance the imaging or therapeutic performance of the radiopharmaceutical. The downside of the pharmacological activity is that receptor binding also results in transmission of a secondary intracellular message which has some biological consequence. Depending on the nature of the biological effect, it may or may not be an acceptable side-effect of the Nuclear Medicine procedure. If it is considered unacceptable then either doses below the pharmacological limit must be used (if possible) or a peptide with antagonist actions be employed, even if these lack the desirable property of internalisation.
2.2 Ligands that map metabolic pathways
A characteristic of tumour cells is their enhanced and uncontrolled rate of proliferation. This rapid growth imposes considerable energy demands upon the cell and results in up-regulation of many transport and synthetic mechanisms including those responsible for metabolism of sugars, amino-acids and nucleotides. The paradigm for ligands that exploit this up-regulation of transporters is glucose. Fluorine-18 labelled deoxyglucose (18FDG) shows enhanced levels of uptake in many tumour types and its clinical application for staging of the disease is rapidly expanding.20 An example is shown in Fig. 2. However, increasing attention is being paid to the effect upon this and other transport mechanisms during treatment of the disease. It is well recognised that much cancer chemotherapy is highly toxic and its efficacy variable, however the effect, or lack of it, only becomes apparent after some time when the tumour either does or does not shrink. Many patients therefore undergo extended courses of such therapy with little benefit. A means of assessing therapy much earlier in the treatment regimen would be very valuable. Early evidence is suggesting that tumours that show a reduction of 18FDG uptake after the first course of chemotherapy will go on to show a benefit from the treatment while those that do not, will not benefit.21 Radioligands that interact with other pathways of cell metabolism are also being explored to see if they provide complimentary information to that obtained with 18FDG. A list of some of the more promising is given in Table 2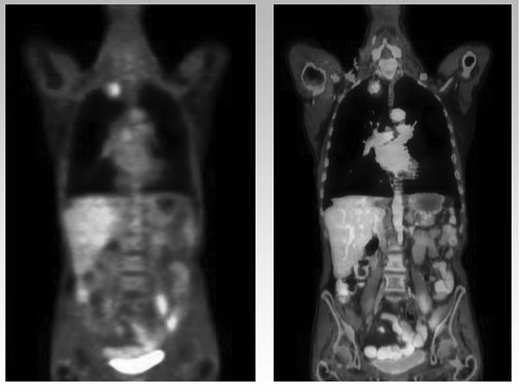 | ||
| Fig. 2 An example of a Nuclear Medicine PET–CT scan using 18FDG to image a patient with lung cancer. On the left a functional 18FDG image of the chest and pelvis showing the ‘hot-spot’ caused by a tumour in the upper quadrant of the right lung. On the right the radionuclide image is fused with a CT image showing the detailed anatomy of the patient. (Figure courtesy of Dr Norbert Avril, Director PET Centre, St Bartholomew's Hospital, London). | ||
![Chemical structures of radioligands listed in Table 2. From top counter-clockwise: A: 2-[18F]fluoro-2-desoxy-d-glucose (18F-FDG), B: 3′-[18F]fluoro-3′-deoxythymidine (18F-FLT), C: 1H-1-(3-[18F]fluoro-2-hydroxypropyl)-2-nitroimidazole (18F-FMISO), D: -[18F]fluoroethyl)-l-tyrosine (18F-FET), E: Caspase-3 substrate 131I-YDEVDG-NH2.](/image/article/2007/MB/b611736h/b611736h-f3.gif) | ||
| Fig. 3 Chemical structures of radioligands listed in Table 2. From top counter-clockwise: A: 2-[18F]fluoro-2-desoxy-D-glucose (18F-FDG), B: 3′-[18F]fluoro-3′-deoxythymidine (18F-FLT), C: 1H-1-(3-[18F]fluoro-2-hydroxypropyl)-2-nitroimidazole (18F-FMISO), D: -[18F]fluoroethyl)-L-tyrosine (18F-FET), E: Caspase-3 substrate 131I-YDEVDG-NH2. | ||
| Pathway | Associated process | Ligand(s) | Reference |
|---|---|---|---|
| GLUT transporters | Glycolysis | 18FDG | 22 |
| Amino acid transporters | Protein synthesis | Aminoacid analogues | 23 |
| Thymidine kinase | Proliferation | Nucleoside | 24 |
| Reduction | Hypoxia | Nitroimidazoles | 25 |
| Caspase enzymes | Apoptosis | Caspase inhibitors | 26 |
3. Radionuclides used in nuclear medicine
A list of the most widely used radionuclides for imaging and therapy is shown in Table 3. The choice of the radionuclide will depend upon the purpose to which the radioligand is being used and will depend upon the physical decay properties of the isotope. Imaging procedures require highly penetrating radiation such as γ-photons or X-rays that can pass through overlying body tissues and are detected by suitable instruments outside the body. Of particular importance for diagnostic procedures is the balance between the clinical value of the information obtained from the procedure and the radiation dose received by the patient. A choice must be made between a radionuclide suitable for either Positron Emission Tomography (PET) or Single Photon Emission Computed Tomography (SPECT) imaging and a decay half-life which matches the pharmacokinetics of the ligand under consideration. For agents with rapid delivery and clearance times, half lives of minutes or hours are preferred while for those with more extended pharmacokinetics , radionuclides that decay with half-lives of several days may be required. Although the requirements of an ideal radionuclide for diagnostic imaging are well understood, this is not the case for therapeutic applications. Non-penetrating radiation, typically β-particle radiation is normally used but recently experimental clinical studies with α-emitting radionuclides have been performed.27 The radiation should interact with the tissues around the site of decay in order to deposit as much energy in the target as possible in order to damage the diseased cells, but should, ideally, have limited effect on normal more distant tissues. It is recognised that the biological effects achieved will depend upon the nature of the particulate emission, its energy and physical half-life and the clearance times of the radiopharmaceutical, however we are far from understanding the best combination of these factors for optimal therapy. The emphasis in the majority of therapeutic studies has been on efficacy in clinical conditions or in animal models of human cancers and very little consideration has been given to the effects of targeted radioactivity at the cellular and molecular level. Despite its undoubted importance, little attention has been paid to radiobiology in therapeutic nuclear medicine. Extrapolations have often been made from the beneficial and toxic effects of external beam radiation despite profound differences in the nature of the radiation, the rate at which it is administered and the site of its action in tissue. Radiation doses to tissues may be averaged over large volumes even though the heterogeneous nature of the activity distribution is well recognised. The cytotoxic effects of radiation arise from the interaction of short-lived reactive chemical species generated by ionisation of the cellular environment with a variety of structural and functional biomolecular structures in the cell. Numerous radiobiological studies have emphasised the importance of DNA as a target for ionising radiation, but the cascade of effects subsequent to the direct or indirect interaction of the radiation with DNA is enormously complex.28 The effect of DNA repair mechanisms is crucial. Although these can effectively correct the damage imposed by radiation, inherent errors in the repair process can have the effect of magnifying the initial lesion from a sub-lethal to a lethal one. In such a situation the relationships between dose-rate, cell growth kinetics, and the period of irradiation will be crucial. Such considerations emphasise the need for an understanding of the effects at the molecular level of the (ultra)-low-dose rate over extended irradiation times delivered by targeted radiation but such data are as yet, sparse.| Radionuclide | Application | Half-life | Main radioactive emissions |
|---|---|---|---|
| Technetium-99 m | Diagnostic SPECT | 6 h | 140 keV gamma |
| Indium-111 | Diagnostic SPECT | 2.8 days | 171 and 245 keV gamma |
| Fluorine-18 | Diagnostic PET | 110 min | 511 keV gammas |
| Gallium-68 | Diagnostic PET | 68 min | 511 keV gammas |
| Yttrium-90 | Therapeutic | 64 h | 2.3 MeV beta |
| Iodine-131 | Therapeutic | 8 days | 606 keV beta, 364 keV gamma |
| Lutetium-177 | Therapeutic | 6.7 days | 497 keV beta, 208 keV gamma |
4. Radiolabelling strategies
The main task of the radiochemist is to develop a means of labelling a ligand with an interesting biological function with the preferred radionuclide in such a way that it does not interfere with its targeting function. The radioisotope must also remain associated with the ligand even after administration into the biological milieu so that the distribution of the radioactivity can be considered to map the distribution of the biological pathway of interest. However, it should also be recognised that the method of labelling can have a profound impact upon the pattern of biodistribution of the radioligand. This biodistribution will be influenced by what may be termed ‘specific’ and ‘non-specific’ mechanisms. Specific mechanisms relate to the targeting property of the ligand for which it has been chosen, for example binding to a particular receptor. Provided it has access, the radioligand will bind to receptors present on either diseased or normal tissues so for example ligands based on gastrin will bind to gastrin receptors present in both tumours and in the stomach. However, the radioligand may also undergo a host of other non-receptor mediated interactions depending on its physico-chemical characteristics. This non-specific behaviour may well play a greater influence on the overall pattern of biodistribution than the specific component. While those factors that influence the receptor-mediated interactions are relatively well understood and can also be studied in vitro, those responsible for the non-specific component are less well defined and can only really be assessed in vivo. As described above, the biodistribution of the perfect (but non-existent) radioligand would reflect only the specific component. The influence of the non-specific component depends upon whether a diagnostic or therapeutic approach is being employed. For diagnosis, its effect may be to mask areas of specific uptake. A good example of this is the case of radiolabelled monoclonal antibodies. Since these large molecules remain in the blood stream for hours or even days after injection, tumours which are close to major blood vessels (such as in the chest for example) will be hard to image. The route of excretion of the radioligand also makes an important contribution to the non-specific component of the biodistribution. Ideally, the tracer will be excreted by the kidneys. This route of excretion is rapid and the kidneys are well-defined organs that are easy to identify on an image. Patients can also be encouraged to void radioactivity from the bladder by urination. If the radioligand is excreted via the hepatobiliary and gastrointestinal tract however, then the excretion is much slower with transit times greater than one day. The structure of the intestines is much less clearly defined on images than the kidneys and this can make it very difficult to distinguish between specific uptake in a tumour in the abdomen and non-specific uptake in the intestines.For therapeutic applications of radioligands the effect of non-specific uptake is normally to increase the radiation dose to normal tissues. This limits the total amount of radioactivity that can be administered for the procedure and therefore the radiation dose that can be delivered to the tumour and the likely success of the procedure.29
The parameters that determine the non-specific component of the biodistribution are understood to some extent. Blood circulation times are determined partly by size so that proteins which are above the cut-off for glomerular filtration by the kidney (about 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) have much longer circulation times than those below this size. Compounds which are lipophilic are likely to be excreted by the hepatobiliary tract and also to show greater levels of binding to serum proteins. Both are undesirable. In this regard hydrophilic compounds are preferred since they show lower protein binding and a propensity for renal excretion,30 however hydrophilic radioligands will pass less easily through the cell membrane so for intracellular targets there may well be a trade-off between achieving a desirable non-specific component to the biodistribution and getting the ligand to its site of action.
000 Da) have much longer circulation times than those below this size. Compounds which are lipophilic are likely to be excreted by the hepatobiliary tract and also to show greater levels of binding to serum proteins. Both are undesirable. In this regard hydrophilic compounds are preferred since they show lower protein binding and a propensity for renal excretion,30 however hydrophilic radioligands will pass less easily through the cell membrane so for intracellular targets there may well be a trade-off between achieving a desirable non-specific component to the biodistribution and getting the ligand to its site of action.
5. Tools for radioligand development
Typically development of a new radioligand will proceed through a number of phases. The initial ‘Chemical’ phase is concerned with the synthesis of the ligand, development of a radiolabelling approach and assessment of purity and stability of the radioligand. The most valuable tool at the stage is radio-HPLC. This technique can be used to measure radiolabelling efficiency, radiochemical purity and changes in this parameter with time and under the influence of ambient conditions. Since most radioligands are present at very low (nM–pM) concentrations, mass spectrometry has in the past struggled to make an impact but recent developments suggest that this may prove to be an increasingly valuable analytical technique.31 The second ‘biological’ phase looks at the interaction of the radioligand with its target in vitro. This can be done using cell cultures or isolated purified cell membranes or receptor preparations. This stage seeks to predict the specific component of the subsequent biodistribution by measuring the affinity of target-ligand interactions, the degree of internalisation and the fate of the internalised radioactivity. To some extent the non-specific component of the biodistribution can also be predicted in vitro by measuring the protein binding and lipophilicity of the radiotracer.32 The final pre-clinical stage is to assess the biodistribution of the radioligand in an animal model. This can be performed by injection into groups of animals followed by killing the animals at selected time points and measuring the levels of radioactivity in resected tissues of interest. Specialised PET or SPECT small animal imaging cameras allow repeated assessment of biodistribution to be made over time using smaller numbers of animals. An example of such an image is shown in Fig. 4.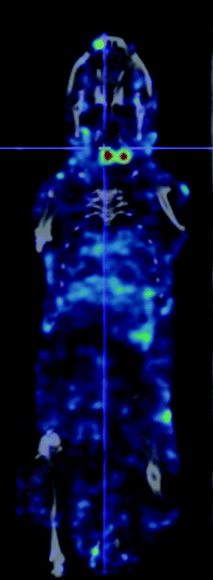 | ||
| Fig. 4 An example of a small animal SPECT/CT scan showing antigen specific imaging of the thyroid gland, by fusing the image of binding of an 125I-labeled anti-TSH receptor antibody to the TSH receptor in the two lobes of the mouse thyroid gland (in red) with a CT-image (in grey) showing the location of the gland in the neck of a normal mouse. (The study was performed in compliance with the UK Animals (Scientific Procedures) Act 1986 and the Code of Practice for the Housing and Care of Animals used in Scientific Procedures (Home Office, UK) with approval of the Animals Ethics committees of Cancer Research UK and Queen Mary, University of London). | ||
7. Concluding remarks
Radiopharmaceutical design is a rewarding field for research, lying as it does on the interface of several different scientific disciplines. The radioligands so developed can usefully be used to answer biological questions in animal models or to assist in the management of patients with a number of life-threatening diseases. The rules which determine the success or otherwise of a radiopharmaceutical design strategy are gradually becoming clearer and in combination with progress in cell and molecular biology will result in exciting developments in this field in forthcoming years.Acknowledgements
The financial support of Cancer Research UK is gratefully acknowledged.References
- S. M. Larson, Cancer Res., 1990, 50, 892s CAS.
- R. K. Jain, Cancer Res., 1990, 50, 814s CAS.
- G. R. Mirick, B. M. Bradt, S. J. Denardo and G. L. Denardo, Q. J. Nucl. Med. Mol. Imaging, 2004, 48, 251 Search PubMed.
- J. S. Huston and A. J. George, Hum. Antibodies, 2001, 10, 127 Search PubMed.
- D. Schrama, R. A. Reisfeld and J. C. Becker, Nat. Rev. Drug Discovery, 2006, 5, 147 CrossRef CAS.
- L. J. Holt, C. Herring, L. S. Jespers, B. P. Woolven and I. M. Tomlinson, Trends Biotechnol., 2003, 21, 484 CrossRef CAS.
- H. K. Binz, P. Amstutz and A. Pluckthun, Nat. Biotechnol., 2005, 23, 1257 CrossRef CAS.
- A. A. Kortt, O. Dolezal, B. E. Power and P. J. Hudson, Biomol. Eng., 2001, 18, 95 Search PubMed.
- J. C. Reubi, Endocr. Rev., 2003, 24, 389 Search PubMed.
- W. A. Breeman, M. de Jong, D. J. Kwekkeboom, R. Valkema, W. H. Bakker, P. P. Kooij, T. J. Visser and E. P. Krenning, Eur. J. Nucl. Med. Mol. Imaging, 2001, 28, 1421 CrossRef CAS.
- M. Bruehlmeier, E. G. Garayoa, A. Blanc, B. Holzer, S. Gergely, D. Tourwe, P. A. Schubiger and P. Blauenstein, Nucl. Med. Biol., 2002, 29, 321 CrossRef CAS.
- M. De Jong, B. F. Bernard, E. De Bruin, A. Van Gameren, W. H. Bakker, T. J. Visser, H. R. Macke and E. P. Krenning, Nucl. Med. Commun., 1998, 19, 283 CAS.
- C. J. Smith, W. A. Volkert and T. J. Hoffman, Nucl. Med. Biol., 2005, 32, 733 CrossRef CAS.
- P. B. Johnston, C. Bondly and I. N. Micallef, Expert Rev. Anticancer Ther., 2006, 6, 861 Search PubMed.
- K. A. Chester, A. Mayer, J. Bhatia, L. Robson, D. I. Spencer, S. P. Cooke, A. A. Flynn, S. K. Sharma, G. Boxer, R. B. Pedley and R. H. Begent, Cancer Chemother. Pharmacol., 2000, 46, S8 CrossRef CAS.
- R. M. Reilly, R. Kiarash, J. Sandhu, Y. W. Lee, R. G. Cameron, A. Hendler, K. Vallis and J. Gariepy, J. Nucl. Med., 2000, 41, 903 CAS.
- M. I. Milowsky, D. M. Nanus, L. Kostakoglu, S. Vallabhajosula, S. J. Goldsmith and N. H. Bander, J. Clin. Oncol., 2004, 22, 2522 CrossRef CAS.
- A. J. Beer, R. Haubner, M. Goebel, S. Luderschmidt, M. E. Spilker, H. J. Wester, W. A. Weber and M. Schwaiger, J. Nucl. Med., 2005, 46, 1333 CAS.
- H. H. Boersma, B. L. Kietselaer, L. M. Stolk, A. Bennaghmouch, L. Hofstra, J. Narula, G. A. Heidendal and C. P. Reutelingsperger, J. Nucl. Med., 2005, 46, 2035 CAS.
- R. Kumar, M. R. Nadig and A. Chauhan, Expert Rev. Anticancer. Ther., 2005, 5, 1079 Search PubMed.
- N. E. Avril and W. A. Weber, Radiol. Clin. North Am., 2005, 43, 189 Search PubMed.
- N. Avril, J. Nucl. Med., 2004, 45, 930 CAS.
- O. Couturier, A. Luxen, J. F. Chatal, J. P. Vuillez, P. Rigo and R. Hustinx, Eur. J. Nucl. Med. Mol. Imaging, 2004, 31, 1182 CAS.
- L. B. Been, A. J. Suurmeijer, D. C. Cobben, P. L. Jager, H. J. Hoekstra and P. H. Elsinga, Eur. J. Nucl. Med. Mol. Imaging, 2004, 31, 1659 CrossRef.
- J. R. Ballinger, Semin. Nucl. Med., 2001, 31, 321 Search PubMed.
- C. Bauer, U. Bauder-Wuest, W. Mier, U. Haberkorn and M. Eisenhut, J. Nucl. Med., 2005, 46, 1066.
- D. Mulford, D. Scheinberg and J. Jurcic, J. Nucl. Med., 2005, 46, 199s–204s.
- J. Pouget and S. Mather, Eur. J. Nucl. Med., 2001 Apr, 28(4), 541–61 Search PubMed.
- M. Melis, E. P. Krenning, B. F. Bernard, R. Barone, T. J. Visser and M. de Jong, Eur. J. Nucl. Med. Mol. Imaging, 2005, 32, 1136 CrossRef CAS.
- M. Schottelius, F. Rau, J. C. Reubi, M. Schwaiger and H. J. Wester, Bioconjugate Chem., 2005, 16, 429 CrossRef CAS.
- W. E. Greenland and P. J. Blower, Bioconjugate Chem., 2005, 16, 939 CrossRef CAS.
- C. Decristoforo and S. J. Mather, Nucl. Med. Biol., 1999, 26, 389 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |