A microfluidic system in combination with optical tweezers for analyzing rapid and reversible cytological alterations in single cells upon environmental changes†
Emma
Eriksson
a,
Jonas
Enger
a,
Bodil
Nordlander
b,
Nika
Erjavec
b,
Kerstin
Ramser
a,
Mattias
Goksör
a,
Stefan
Hohmann
b,
Thomas
Nyström
b and
Dag
Hanstorp
*a
aDepartment of Physics, Göteborg University, SE-412 96, Göteborg, Sweden. E-mail: dag.hanstorp@physics.gu.se; Fax: +46 31 772 2092; Tel: +46 31 772 3274
bDepartment of Cell and Molecular Biology, Göteborg University, Box 462, SE-405 30, Göteborg, Sweden
First published on 4th October 2006
Abstract
We report on the development of an experimental platform where epi-fluorescence microscopy and optical tweezers are combined with a microfluidic system to enable the analysis of rapid cytological responses in single cells. The microfluidic system allows two different media to be merged in a Y-shaped channel. Microscale channel dimensions ensure purely laminar flow and, as a result, an environmental gradient can be created between the two media. Optical tweezers are used to move a single trapped cell repeatedly between the different environments. The cell is monitored continuously by fluorescence microscopy during the experiment. In a first experiment on yeast (Saccharomyces cerevisiae) we observed changes in cell volume as the cell was moved between environments with different osmolarity. This demonstrated that the platform allowed analysis of cytological alterations on a time scale shorter than 0.2 s. In a second experiment we observed the spatial migration of the Yap1p transcription factor fused to GFP as a cell was moved from an environment of low to high oxidative capacity. The system is universal allowing the response to numerous environmental changes to be studied on the sub second time scale in a variety of model cells. We intend to use the platform to study how the age of cells, their progression through the cell cycle, or their genetic landscape, alter their capacity (kinetics and amplitude) to respond to environmental changes.
Introduction
Microfluidic systems are becoming an increasingly important tool for chemical, biochemical and biological analysis, as illustrated by the large number of publications in recent years.1,2 Microfluidic systems (or lab-on-a-chip devices) generally refer to channel systems where at least one dimension is on the order of microns. Microfluidic systems are sometimes described simply as miniaturizations of macroscale systems. Although such a description applies in some cases, several new phenomena occur upon scaling down to micron dimensions. For instance, the surface-to-volume ratio is dramatically increased and the flows created in such systems are almost exclusively laminar.1,3,4 By making use of these phenomena new types of devices are made possible, and enable experiments that would otherwise be impossible to perform.The most commonly used method of fabricating microfluidic systems is soft lithography, where the channel systems are made of a rubber silicon, such as poly(dimethylsiloxane), PDMS.5 This material has a number of useful properties; it is optically transparent, non-toxic to cells, its surface properties can be modified and it is impermeable to water but permeable to gases.6
In a microfluidic system the flow is typically laminar, with Reynolds numbers below 1. Consequently, fluids from two channels that merge into a single channel will flow in adjacent separate laminar streams and mix only by diffusion.6 The concentration gradient between two such flows has been characterized both experimentally and theoretically,7–9 and channel designs have been developed to tailor the profile of the gradient.10 There are also schemes that use parallel laminar flows in microchannels for diffusion based separation and detection.11 Separation in microfluidic systems has also been achieved using optical fields.12
Optical tweezers are a tool used to manipulate biological objects like cells, bacteria and viruses under the microscope.13,14 The principles of optical tweezers (or single beam gradient force optical trap) were first described by Ashkin,15 who also showed that it was possible to trap micrometer-sized particles in solution. Proper choice of the trapping wavelength minimizes absorption and photo-damage in trapped biological objects.16,17 Near infrared light has proven to be most suitable. Since the cell is trapped without physical contact it is exposed to solution from all sides. Hence, adsorption-induced effects can be avoided and the environment can be kept contamination-free. Optical tweezers have found widespread use within the field of cell biology for single cell experiments.18
Information on how cells respond to changes in their external or internal environment, interact with each other, or undergo complex processes such as differentiation, is commonly gained from the analysis of large populations or ensembles of cells. Recent advances in technology, including optical tweezers, microfluidics and advanced imaging techniques, help reveal information at a single cell level. Such information will help determine to what extent differences between individual cells (stochastic noise) may give rise to non-deterministic behaviour at the population level. Parallel analysis of living single cells can also uncover dynamics and individual phenotypic variations that would be missed in experiments at the population level. Hence, single cell studies will provide new insight into the system level properties of signalling pathways and their dependence on cellular age, degree of development, and cell cycle progression.19
In order to carry out biological experiments in microchannels, cells are commonly immobilized at the bottom of the channel, or carried by the flow. If the cell is immobilized in a channel junction it is possible to treat the cell locally with different chemicals. Such systems where the cell is immobilized on the bottom of the channel have been used to deliver small molecules to selected domains inside the cell20 and to study lateral propagation of epidermal growth factor signalling.21 The same kind of microfluidic system has also been used to study the embryonic development of a fruit fly in a temperature step.22,23 Schilling et al. have constructed a microfluidic device for cell lysis and fractionation/detection of intracellular components.24
When performing single cell studies it is often important to control the movement of the cell in the microfluidic system. One solution is to trap the cell hydrodynamically in a 3-D microfluidic system.25,26 The trap is formed by creating points of stagnation, where the fluid speed is zero. With such a system, Wheeler et al. were able to perform one complete solution change for the cell in approximately 100 ms.25 Other similar approaches involve guiding the cell by a controlled flow into a U or V-shaped cell retention structure where it can be kept during the study.27,28 Another method is to hold the cell with a micropipette. Combined with microfluidics, this technique has been used to scan a patch-clamped cell across discrete zones of different solutions created in an open volume.29,30 The exposure times for the cell to each environment could be as short as a few milliseconds. Other techniques for controlling the position of a cell in a microfluidic system include constructing physical “obstacles” to stop the cell31 and dielectrophoretic trapping.32
A different approach is to use optical tweezers to hold and manipulate the cell within the microfluidic system. Wang et al. did early work on combining microfluidic systems and optical tweezers.33 More recently, optical tweezers have been used to position bacteria in a single-cell microcultivation assay,34 to transport cells in a microfluidic system for subsequent lysis of cells and recording of electropherograms,35 and combined with microfluidics to observe rotational motion of cells.36 Munce et al. used optical tweezers to pick and transport cells that were subsequently analyzed individually by capillary electrophoresis in parallel microchannels.37
A challenge in single cell studies is to rapidly and completely change the medium around the cell. This is of interest because cellular responses may occur on a sub-minute time scale. The question was addressed in our previous work, where we demonstrated how cells can be moved with the optical tweezers between different reservoirs containing different media, without the media being dragged along with the trapped cells.38 A limitation with this system was that the different media were contained in separate reservoirs that were connected by relatively long channels. The time needed to change the medium around the cell was thus limited by the time required to transport the cell between the different reservoirs, which was on the order of a minute. In this paper we show that it is possible to reversibly change the medium around a cell within a fraction of a second by utilizing optical tweezers and laminar flow in a Y-shaped microfluidic system, while monitoring the cell with various imaging techniques. We demonstrate the power of this system by measuring cell volume changes in yeast (Saccharomyces cerevisiae) upon changes in medium osmolarity and localization of a transcription factor upon changes in oxidative stress.
Method
Microfluidic system
The microfluidic system is fabricated from rubber silicon (Sylgard 184, Dow Corning, USA). The technique is described in detail elsewhere,38 and is only described briefly here. The channel design is transferred with e-beam lithography to a chromium mask, which is used to create a positive relief of the channel structures in SU-8 25 photoresist. A PDMS cast of the channel system is sealed to a cover slip and tubing connectors are attached to the inlet holes to allow a flow to be established by a syringe pump.A schematic of the channel design is shown in Fig. 1a. The two inlet channels are 50 µm wide and the combined channel is 100 µm wide. The channels are 27 µm high (measured with a profilometer). Flows in the inlet channels are typically around 40 nl min−1. The characteristic length scale, L, of the system can be estimated using the hydraulic diameter 4A/P = 43 µm, where P is the wetted perimeter and A is the cross sectional area of the principal channel. This implies a Reynolds number (Re = ULρ/η) of 0.02 (with the typical flow velocity, U, taken as the average flow velocity at pumping rate 40 nl min−1), using the density, ρ, and viscosity, η, of water. This assures laminar flow behaviour.
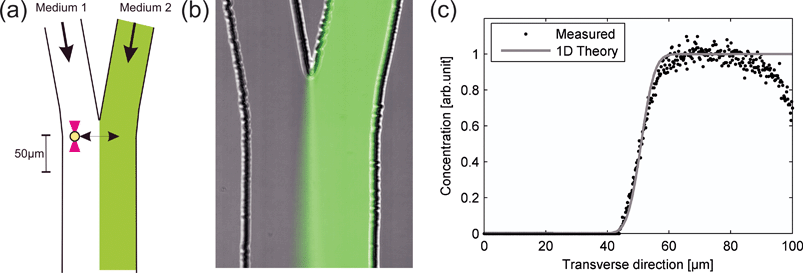 | ||
| Fig. 1 (a) A schematic drawing of the microfluidic system showing how a trapped cell can be reversibly moved between two different media. (b) A microscope image, showing the microfluidic system when fluorescein has been added to the medium in the right channel. (c) Measurement of the fluorescein concentration profile together with a theoretical expression 10 µm downstream from the point where the two flows meet. | ||
The principle of creating two closely spaced environments is shown in Fig. 1b, where, for visualization purposes, fluorescein was added to the right channel. The corresponding concentration profile extracted from the microscope image (assuming a linear relationship between fluorescence intensity and fluorescein concentration) 10 µm downstream from the position where the two flows meet at a pumping rate of 40 nl min−1 is shown in Fig. 1c, together with a 1-D theoretical expression39
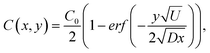
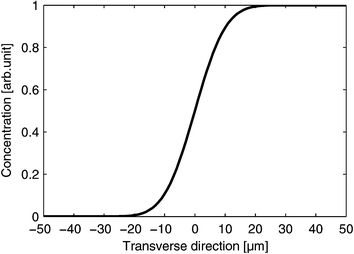 | ||
| Fig. 2 Theoretical estimation of the NaCl concentration profile 10 µm downstream at an average flow of 40 nl min−1. | ||
Microscope
Imaging was performed using a Nikon TE300 inverted microscope equipped with an epi-fluorescence excitation system including a 100 W mercury lamp for excitation and a CCD camera for detection. The microscope was equipped with an oil immersion Nikon Plan Fluor 100X objective with a numerical aperture of 1.3. The microscope was also equipped with a programmable motorized xy stage (repeatability <1 µm and resolution 0.02 µm).Optical tweezers
The design of the optical tweezers setup has been described elsewhere41 and is only described briefly here. A schematic of the setup is shown in Fig. 3. An ytterbium fiber laser with a wavelength of 1070 nm and a collimated output of diameter 5.2 mm was coupled into the microscope through a telescope (lenses L1 and L2). Lens L1 was used to adjust the trapping plane, such that it coincided with the image plane of the microscope objective. The laser power measured at the back aperture of the microscope objective was 360 mW, indicating that 220 mW was used for trapping.42 A trapped object can be moved within the microfluidic system using the motorized microscope stage to change the position of the channel system relative to the fixed trap. The motion control is best in the directions perpendicular to the optical axis (i.e. in the image plane), while the distance the cell can be moved in the direction of the optical axis is more limited. However, in practice, this is not a limitation since we are working with a planar microfluidic system. The principle of how the optical tweezers are used to move the cell within the microfluidic system is illustrated in Fig. 1a.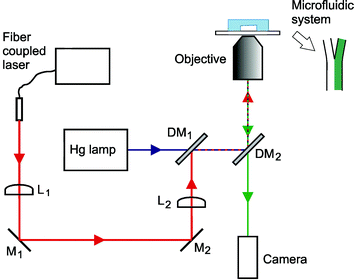 | ||
| Fig. 3 Experimental setup. The output of the fiber coupled laser used for the optical tweezers was directed through the microscope objective, by the use of a telescope (lenses L1 and L2), mirrors (M1 and M2) and dichroic mirrors (DM1 and DM2). The first dichroic mirror allows the excitation light from the mercury lamp to pass through, while reflecting the trapping laser light into the beam path. The second dichroic mirror reflects the excitation light and trapping laser, while transmitting the visible light for imaging. The microfluidic system is placed on the microscope stage. | ||
The time required to establish the environmental change depends, in addition to the steepness of the concentration gradient, on how fast we can move the cell. This, in turn, depends on the strength of the optical trap. By increasing the laser power, however, one faces the risk of inducing photo-damage in the cell. Any experiment using the optical tweezers therefore requires control experiments where one assures that the laser intensity experienced by the cell is kept below the damage threshold.
Image analysis
Image analysis of changes in yeast cell volume during salt stress was performed using a custom-written program in MATLAB. The program estimates the yeast cell area from the microscope images based on edge detection using a Sobel filter and the morphological operations dilation followed by erosion. This area is then converted to a volume by assuming that the volume change is isotropic.Results
Two different experiments were performed to show the functionality of the experimental platform. First, yeast cells (S. cerevisiae) were exposed to salt stress, by capturing individual cells from the channel containing standard yeast growth medium (YNB, yeast nitrogen base) and moving them to the adjacent channel which in addition contained sodium chloride. When exposed to an environment of lower water activity, water flows out of the cell due to osmosis, causing the cell to shrink. We have monitored the size of the yeast cell while moving it back and forth repeatedly between the two environments. By studying the behaviour of single yeast cells during osmotic shock (single shock or multiple shocks) one will gain more insight into how the high osmolarity glycerol (HOG) signalling pathway, a very well-studied mitogen-activated protein (MAP) kinase system, is regulated.43The change of the volume of a single yeast cell following successive osmotic shocks, as estimated from the microscope images, is shown in Fig. 4. The volume is normalized such that the initial volume is equal to 1. The yeast cell was initially captured with the optical tweezers in the channel containing yeast growth medium. The cell was then moved across the boundary between the two flows to a medium with 1 M NaCl. In the saline environment the cell was kept for 16 s before it was moved back to the non-saline flow. This movement back and forth between the two flows was controlled by the programmable microscope stage and was performed three times. The reversible response of the cell is demonstrated in Fig. 4a. In the experiment the cell was moved a distance of 80 µm with a speed of 500 µm s−1, thus the total time used to move the cell was less than 0.2 s. Depending on the width of the gradient zone, the change of environment can be even quicker. In Fig. 4b we can see that the flow of water into the cell starts within 0.2 s.
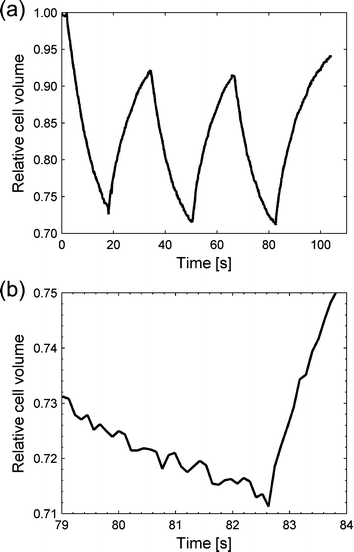 | ||
| Fig. 4 NaCl stress of a yeast cell. (a) The estimated yeast cell volume from the microscope images shown over time. The cell started in the left “neutral” flow, and soon after the recording was started, was moved to the saline flow where water started to flow out from the cell. After a while the cell was moved back to the first flow and water flowed back in. The procedure was performed three times. (b) A close-up view of the graph when moving back to the “neutral” flow the third time. The inflow of water started within 0.2 s. | ||
The goal with the second experiment was to show that our experimental platform also allows for studies over long time scales while continuously imaging the cell. This was done by exposing yeast cells to 5 mM tert-butyl hydroperoxide (t-BOOH) stress. During the whole process it was possible to avoid surface adhesion of the cell while keeping it in fresh media. The yeast cells had a green fluorescent protein (GFP) fused to the Yap1 protein, which is a transcription factor involved in the cell's response to an oxidative stress.44 Initially the Yap1p-Gfp proteins were spread over the cytosol, but upon peroxide stress the proteins accumulated in the nucleus to induce an appropriate transcriptional response in order to counteract the stress.
In the experiment we moved the yeast cell to the t-BOOH environment, and kept it there for about 30 min before moving it back to the “neutral” environment, while imaging the cell with the epi-fluorescence microscope. A transmission image of the cell at the beginning of the experiment is shown in Fig. 5a. The accumulation of fluorescent GFP in the nucleus was monitored and is shown in Fig. 5b and 5c, at the start of the experiment and after 27 min in the peroxide environment, respectively. Back to the “neutral” environment it was possible to observe how the cell downregulated the stress response by returning to normal spatial distribution of GFP. The image in Fig. 5d was taken 20 min after moving back to the flow with pure growth medium.
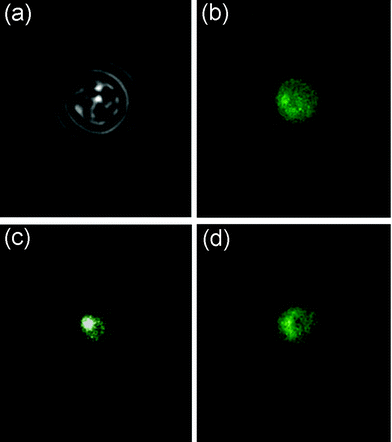 | ||
| Fig. 5 t-BOOH stress of a yeast cell. The cell started in the left “neutral” channel. A transmission image of the cell is shown in image (a). The Yap1p-Gfp proteins are distributed over the entire cytosol, shown in image (b). The cell was then moved to the t-BOOH environment upon which the Yap1p-Gfp accumulated in the nucleus, which can be seen in image (c) that is taken after 27 min. The intensity in the white spot is much higher than in the green parts. The cell was then moved back to the “neutral” environment, where the spatial GFP distribution returned to normal. Image (d) is taken after 20 min of recovery. | ||
Discussion
A general trend in microbiology is to move from experiments on populations of cells to studies on single cells. For instance, detailed knowledge on cellular signalling pathways can be achieved by studying changes in the localization of a fluorescent protein in a cell as it is exposed to different media. There are many requirements on the experimental platform of such an experiment. It should offer rapid, reversible and controlled environmental changes, the entire cell should be exposed to the media without any contamination, it should be possible to select a specific cell from a large population and it should be possible to repeat the experiment on a different cell under identical conditions. Further, parallel single cell experiments are highly requested to allow investigations of a large number of cells on an acceptable time scale. The system must also allow various techniques for registration of the cell response. Last, but not least, the technique used to manipulate the cell must be non-invasive for the cell under investigation.A major breakthrough in single cell studies came with the introduction of the lab-on-a-chip concept, which allowed cells to be kept in controlled environments while continuously being observed using advanced imaging techniques. In previous lab-on-a-chip based methods several different approaches for manipulation of the cells have been used.
Several groups have used the method to immobilize the cell either on the bottom or towards micro-fabricated obstacles in the microfluidic system. These methods are simple since no external means are necessary to control the cell position. This is an advantage, but it also imposes experimental constraints. The system is easy to operate, but the flexibility in controlling the cell position is limited. Further, the interaction with the surface might affect the cell, and for repeated experiments it is hard (or impossible) to dispose of old cells. This limits the number of cells that can be investigated with each microfluidic system.
An alternative method to fix the cell in space is to create stagnation points in the flow. This allows the media to get in contact with the entire cell surface, and further, the cell can be ejected from the system after completion of the experiment. However, the cell is moved with the medium and hence the total exchange of the environment is less controlled and the speed at which the medium can be changed is limited. Further, in using this method only a few cells can be monitored simultaneously.
By introducing optical tweezers for spatial control of the cells, as has been shown in this paper, these limitations can all be addressed. The entire cell can be exposed to media and it is easy to select a specific cell out of a population in order to position it wherever desired in the system for further studies. The method allows repeated changes between two environments with good time resolution, thanks to the high degree of control over the cell position, which is also independent of the fluid movement. Hence, the cells can rapidly be moved between areas of well defined concentrations of different media. After the experiment, the cell can easily be released, and the system is immediately ready for a new experimental cycle. The drawback is the extra complexity of introducing the optical tweezers into the system. However, they are becoming more and more of a standard tool in the biologist's lab, where they can be combined with most imaging techniques, such as epi-fluorescence, confocal or multiphoton microscopy.45 A further drawback involving optical tweezers is the risk of inducing photodamage to fragile biological samples. However, numerous experiments in biology have shown that this risk can be minimized by a proper choice of the wavelength of the trapping laser. Nevertheless, control experiments should always be conducted to make sure that the laser radiation is not affecting the cells negatively before conclusions from a biological experiment can be made.
Another approach to study rapid environmental changes is to use a micropipette to manipulate a single cell. With this method stronger forces can be applied on the cell, and hence faster environmental changes can be achieved. The main drawback with this technique is, in the same way as when immobilizing cells on a surface, that only part of the cell is exposed to the medium. Furthermore, the method of manipulation also requires an open device to allow the micropipette to be introduced to the system. By using the optical trapping technique we can, but are not limited to, work with a sealed device. A sealed device facilitates keeping the environment contamination-free and also offers more opportunities to control the fluid motion. Another advantage with the optical tweezers approach is that it allows, with only a small increase in the complexity, for investigation of many single cells in parallel by the introduction of holographic optical tweezers.46 With this technique it is possible to split the laser beam into several traps. This possibility to study many cells subjected to the same experimental conditions is an advantage both for obtaining good statistics and for studies comparing different cell types.
Conclusions
With a combination of optical tweezers, microfluidics and microscopy, we present a method that allows the optical image analysis of cellular responses to environmental change taking place in less than 0.2 s. By choosing optical tweezers for manipulation of the cell we have full control over the cell position and the cell environment, since the cell is completely surrounded by the medium and the movement of the cell is separated from the fluid motion. The system presented here enables changing the environment of a single cell quickly and reversibly, while at the same time offering the possibility of monitoring the cell with advanced imaging techniques (also under longer periods of time). This will potentially have important applications within the field of molecular cell physiology. By monitoring a single cell during stress we can get insight into the time constants of different intracellular processes and measure other properties of the cell using appropriate tagging of relevant proteins or monitoring of cell size or shape. We foresee great potential for this system as a general approach for looking at the effects of local environmental changes on single cells. Interesting types of environmental changes that can be investigated are pH-level, osmolarity, ion concentrations, and access to nutrition or potential drugs. In the future, we intend to expand our optical system to include holographic optical tweezers. By doing so, parallel investigation of single cells will be possible.Acknowledgements
This work was supported by the European Commission 6th Framework Programme through the projects ATOM-3D (to DH: Contract No. 508952) and QUASI (to SH: LSH-CT2003-503230), the European Science Foundation EUROCORES Programme SPANAS (to DH) through funds from the Swedish Research Council and from the European Commission 6th Framework Programme, and the SSF-BioX programme (to DH and TN).References
- T. M. Squires and S. R. Quake, Rev. Mod. Phys., 2005, 77, 977–1026 CrossRef CAS.
- A. E. Kamholz, Lab Chip, 2004, 4, 16N–20N RSC.
- D. J. Beebe, G. A. Mensing and G. M. Walker, Annu. Rev. Biomed. Eng., 2002, 4, 261–286 Search PubMed.
- D. R. Reyes, D. Iossifidis, P. A. Auroux and A. Manz, Anal. Chem., 2002, 74, 2623–2636 CrossRef CAS.
- J. C. McDonald, D. C. Duffy, J. R. Anderson, D. T. Chiu, H. K. Wu, O. J. A. Schueller and G. M. Whitesides, Electrophoresis, 2000, 21, 27–40 CrossRef CAS.
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Y. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335–373 Search PubMed.
- A. E. Kamholz, E. A. Schilling and P. Yager, Biophys. J., 2001, 80, 1967–1972 CrossRef CAS.
- A. E. Kamholz, B. H. Weigl, B. A. Finlayson and P. Yager, Anal. Chem., 1999, 71, 5340–5347 CrossRef CAS.
- A. E. Kamholz and P. Yager, Biophys. J., 2001, 80, 155–160 CrossRef CAS.
- S. K. W. Dertinger, D. T. Chiu, N. L. Jeon and G. M. Whitesides, Anal. Chem., 2001, 73, 1240–1246 CrossRef CAS.
- B. H. Weigl and P. Yager, Science, 1999, 283, 346–347 CrossRef.
- M. P. MacDonald, G. C. Spalding and K. Dholakia, Nature, 2003, 426, 421–424 CrossRef CAS.
- A. Ashkin, J. M. Dziedzic and T. Yamane, Nature, 1987, 330, 769–771 CrossRef CAS.
- A. Ashkin and J. M. Dziedzic, Science, 1987, 235, 1517–1520 CrossRef CAS.
- A. Ashkin, J. M. Dziedzic, J. E. Bjorkholm and S. Chu, Opt. Lett., 1986, 11, 288–290 Search PubMed.
- K. Konig, Y. Tadir, P. Patrizio, M. W. Berns and B. J. Tromberg, Hum. Reprod., 1996, 11, 2162–2164 CAS.
- M. Ericsson, D. Hanstorp, P. Hagberg, J. Enger and T. Nystrom, J. Bacteriol., 2000, 182, 5551–5555 CrossRef CAS.
- M. J. Lang and S. M. Block, Am. J. Phys., 2003, 71, 201–215 CrossRef.
- B. F. Brehm-Stecher and E. A. Johnson, Microbiol. Mol. Biol. Rev., 2004, 68, 538–559 CrossRef CAS.
- S. Takayama, E. Ostuni, P. LeDuc, K. Naruse, D. E. Ingber and G. M. Whitesides, Nature, 2001, 411, 1016–1016 CrossRef CAS.
- A. Sawano, S. Takayama, M. Matsuda and A. Miyawaki, Dev. Cell, 2002, 3, 245–257 CrossRef CAS.
- E. M. Lucchetta, J. H. Lee, L. A. Fu, N. H. Patel and R. F. Ismagilov, Nature, 2005, 434, 1134–1138 CrossRef CAS.
- E. M. Lucchetta, M. S. Munson and R. F. Ismagilov, Lab Chip, 2006, 6, 185–190 RSC.
- E. A. Schilling, A. E. Kamholz and P. Yager, Anal. Chem., 2002, 74, 1798–1804 CrossRef CAS.
- A. R. Wheeler, W. R. Throndset, R. J. Whelan, A. M. Leach, R. N. Zare, Y. H. Liao, K. Farrell, I. D. Manger and A. Daridon, Anal. Chem., 2003, 75, 3581–3586 CrossRef CAS.
- X. Y. Peng and P. C. H. Li, Anal. Chem., 2004, 76, 5273–5281 CrossRef CAS.
- P. C. H. Li, L. de Camprieu, J. Cai and M. Sangar, Lab Chip, 2004, 4, 174–180 RSC.
- X. J. Li and P. C. H. Li, Anal. Chem., 2005, 77, 4315–4322 CrossRef CAS.
- J. Sinclair, J. Pihl, J. Olofsson, M. Karlsson, K. Jardemark, D. T. Chiu and O. Orwar, Anal. Chem., 2002, 74, 6133–6138 CrossRef CAS.
- J. Olofsson, J. Pihl, J. Sinclair, E. Sahlin, M. Karlsson and O. Orwar, Anal. Chem., 2004, 76, 4968–4976 CrossRef CAS.
- A. Valero, F. Merino, F. Wolbers, R. Luttge, I. Vermes, H. Andersson and A. van den Berg, Lab Chip, 2005, 5, 49–55 RSC.
- J. Voldman, M. L. Gray, M. Toner and M. A. Schmidt, Anal. Chem., 2002, 74, 3984–3990 CrossRef CAS.
- W. Wang, Y. Liu, G. J. Sonek, M. W. Berns and R. A. Keller, Appl. Phys. Lett., 1995, 67, 1057–1059 CrossRef CAS.
- S. Umehara, Y. Wakamoto, I. Inoue and K. Yasuda, Biochem. Biophys. Res. Commun., 2003, 305, 534–540 CrossRef CAS.
- W. Hellmich, C. Pelargus, K. Leffhalm, A. Ros and D. Anselmetti, Electrophoresis, 2005, 26, 3689–3696 CrossRef CAS.
- J. P. Shelby, S. A. Mutch and D. T. Chiu, Anal. Chem., 2004, 76, 2492–2497 CrossRef CAS.
- N. R. Munce, J. Z. Li, P. R. Herman and L. Lilge, Anal. Chem., 2004, 76, 4983–4989 CrossRef CAS.
- J. Enger, M. Goksor, K. Ramser, P. Hagberg and D. Hanstorp, Lab Chip, 2004, 4, 196–200 RSC.
- P. Tabeling, Introduction to Microfluidics, Oxford University Press Inc., New York, 2005 Search PubMed.
- P. Vanýsek, in CRC Handbook of Chemistry and Physics, Internet Version 2005, ed. D. R. Lide, CRC Press, Boca Raton, FL, 2005 Search PubMed.
- M. Goksor, J. Enger, K. Ramser and D. Hanstorp, Proc. SPIE—Int. Soc. Opt. Eng., 2003, 4966, 50–57.
- K. C. Neuman, E. H. Chadd, G. F. Liou, K. Bergman and S. M. Block, Biophys. J., 1999, 77, 2856–2863 CrossRef CAS.
- S. Hohmann, Microbiol. Mol. Biol. Rev., 2002, 66, 300–372 CrossRef CAS.
- A. Delaunay, A. D. Isnard and M. B. Toledano, EMBO J., 2000, 19, 5157–5166 CrossRef CAS.
- M. Goksor, J. Enger and D. Hanstorp, Appl. Opt., 2004, 43, 4831–4837 CrossRef.
- J. E. Curtis, B. A. Koss and D. G. Grier, Opt. Commun., 2002, 207, 169–175 CrossRef CAS.
Footnote |
| † The HTML version of this article has been additionally enhanced with colour images. |
| This journal is © The Royal Society of Chemistry 2007 |