A high-throughput microfluidic real-time gene expression living cell array†
Kevin R.
King‡
abd,
Sihong
Wang‡
ad,
Daniel
Irimia
ad,
Arul
Jayaraman
c,
Mehmet
Toner
abd and
Martin L.
Yarmush
*abd
aCenter for Engineering and Medicine and Department of Surgery, Massachusetts General Hospital, 51 Blosson St. Rm 406, Boston, MA 02114, USA. E-mail: ireis@sbi.org; Fax: (617) 371-4950; Tel: (617) 371-4882
bMassachusetts Institute of Technology, Division of Health Science and Technology, Boston, MA 02114, USA
cDepartment of Chemical Engineering, Texas A&M University, Boston, MA 02114, USA
dShriners Hospitals for Children, and Harvard Medical School, Boston, MA 02114, USA
First published on 29th September 2006
Abstract
The dynamics of gene expression are fundamental to the coordination of cellular responses. Measurement of temporal gene expression patterns is currently limited to destructive low-throughput techniques such as northern blotting, reverse transcriptionpolymerase chain reaction (RT-PCR ), and DNA microarrays. We report a scalable experimental platform that combines microfluidic addressability with quantitative live cell imaging of fluorescent protein transcriptional reporters to achieve real-time characterization of gene expression programs in living cells. Integrated microvalve arrays control row-seeding and column-stimulation of 256 nanoliter-scale bioreactors to create a high density matrix of stimulus–response experiments. We demonstrate the approach in the context of hepatic inflammation by acquiring ∼5000 single-time-point measurements in each automated and unattended experiment. Experiments can be assembled in hours and perform the equivalent of months of conventional experiments. By enabling efficient investigation of dynamic gene expression programs, this technology has the potential to make significant impacts in basic science, drug development, and clinical medicine.
Introduction
Gene expression dynamics are central to the orchestration of cellular responses. Precisely timed genetic programs coordinate progression through the stages of development,1 responses to metabolic and biosynthetic shifts,2 and adaptations to infectious or environmental stresses.3 Some transcriptional programs are inducible,4 being called upon transiently, only when needed. Others are in constant motion,5 continuously integrating inputs from neighboring cells, neurovascular inputs, and circulating hormones to modulate expression patterns. The ability to monitor transcriptional regulators in real time during physiological and pathological responses would greatly enhance our understanding of cellular regulation and control. Unfortunately, temporal patterns of gene expression remain largely uncharacterized due to the laborious and expensive nature of destructive single-time-point measurement techniques. Here we report a high-throughput experimental platform that allows the dynamic activity of multiple gene regulatory sequences to be monitored in parallel, in living cells, and under many experimental conditions.Conventional gene expression analysis typically involves measuring single-time-point ‘snapshots’ with destructive techniques such as northern blots, reverse transcriptionpolymerase chain reaction (RT-PCR), or DNA microarrays. Using these methods, dynamics can only be approximated by assembling average responses from separate cell populations, one for each time point. While the methods have provided early data on expression dynamics, the laborious and expensive nature of the approaches place significant limitations on both the number of time points and the range of experimental conditions that can be reasonably explored. Recent developments in the area of fluorescent reporter technologies are now permitting nondestructive monitoring of a broad range of intracellular molecular events.
Reporter assays involve transfecting cells with plasmid DNA encoding an easily measured protein such as green fluorescent protein (GFP) under the regulation of a specific transcription factor. When the transcription factor of interest is active, the protein is expressed and GFP levels increase. When the transcription factor is not active, GFP levels decrease. Libraries of transcriptional reporters have been developed in bacteria and used to study regulatory programs involved in biosynthesis6 and flagella assembly;7 however adapting these strategies to adherent mammalian cells is challenging due to low fluorescence signals, stringent growth requirements, heterogeneous morphologies, and continuous cell motion.
To date, high-throughput array technologies have focused significant attention on increasing the number of “outputs” (primarily genes and proteins). However, comparatively little effort has been directed at scaling the number of “inputs,” or cell stimuli. The ability to create complex patterns of soluble stimuli that simulate the dynamic cellular microenvironment in a highly parallel fashion would enable systematic characterization of cell responses and underlying gene regulatory programs. Microfluidics offers an attractive platform for massively parallel integration of cell culture and stimulus control. Microscale fluidic circuits have already proven valuable for automation of biochemical assays such as RT-PCR ,8 and their compatibility with transparent and biocompatible polymers has lead to applications in adherent mammalian cell studies ranging from developmental biology9 and stem cell differentiation10 to mechanotransduction11 and cell migration.12
In this work, we combine microfluidic perfusion culture and molecular stimulation with live cell transcriptional reporter monitoring to create a real-time gene expression array as shown schematically in Fig. 1. Rows of nanoliter-scale bioreactors are seeded with a library of monoclonal reporter cell lines and stimulated by columns of soluble stimuli. The resulting matrix of experiments is noninvasively monitored using time-lapse fluorescence microscopy and quantified using automated image analysis to measure stimulus–response dynamics across a broad range of experimental conditions.
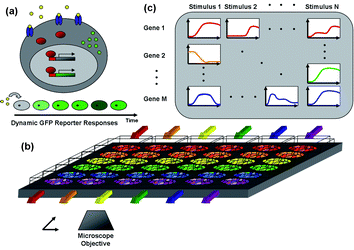 | ||
| Fig. 1 Schematic of the microfluidic real-time gene expression array. (a) Transcriptional reporter operation—when an extracellular stimulus (yellow) binds its receptor (blue) and activates intracellulartranscription factors (red), they enter the nucleus and bind to DNA response elements that activate transcription of the associated genes (grey). In addition to regulating endogenous genes, transcription factors also bind to response elements of the stably transfected reporter plasmid DNA resulting in expression of the reporter protein d2EGFP (green) which can be detected by fluorescence microscopy. If the reporter is not continuously expressed, cellular fluorescence will fade due to the short half-life of the fluorescent protein. (b) Microfluidic multi-reporter array—reporter cell lines for multiple genes and transcription factors are seeded in separate channels of the microfluidic array and stimulated with soluble stimuli in the orthogonal direction (colored arrows). (c) The addressable cellular array is monitored noninvasively by automated time-lapse fluorescence microscopy, and images are quantified by automated image analysis to create a dense 2D matrix of dynamic stimulus–response data. | ||
Guided by our interest in hepatic inflammation,13–15 we created reporter cell lines to monitor the dynamics of several key transcription factors. The library of reporters were seeded in the array and exposed to molecular stimuli commonly encountered during hepatic inflammation—bacterial toxins, cytokines, hormones, and their combinations. In each automated and unattended experiment, we collected 192 time courses, sampled every 90 min for 36 h, totaling ∼5000 single-time point measurements per experiment. Our results revealed distinct dynamics for each pathway and provided evidence for cross-talk between classically independent pathways. In comparison to conventional techniques, which would require months to perform a comparable experiment, the microfluidic real-time gene expression array requires only hours for fabrication and assembly, enabling high-throughput investigation of the coordinated dynamics of gene expression programs.
Results
The microfluidic real-time gene expression living cell array consists of two fundamental components (1) a microfluidic array that allows isolated seeding of multiple reporter cell lines and orthogonal delivery of soluble stimuli, and (2) a library of fluorescent reporter cell lines that dynamically report on the activity of transcription factors of interest.Microfluidic multi-reporter array fabrication and characterization
The microfluidic multi-reporter arrays are constructed using two-layer soft lithography and microstructured membranes .16 A fully assembled device is shown in Fig. 2a. Layer 1 consists of an array of 256 circular ∼10 nl “cell visualization chambers” that can be separated into 64 different experiments by 2 sets of reversible polydimethylsiloxane (PDMS) barriers. The second layer consists of two valve control lines that enable independent manipulation of the reversible barriers, allowing the underlying culture array to be interchanged between isolated rows or columns. The barriers function as “normally closed” valves and prevent cell and fluid communication between adjacent cell culture chambers (Fig. 2b). However, when negative pressure is applied to the appropriate layer 2 control channel by drawing a small amount of fluid into a 1 ml fluid filled syringe, the corresponding barriers can be raised to allow communication between the chambers on either side (Fig. 2c). This reversible barrier design expands upon previously described highly integrated microfluidic arrays17 by making them compatible with mammalian cells. When the barriers are closed, they passively prevent cell movement, yet they can be temporarily opened wide enough (>50 µm) to allow passage of mammalian cells. In doing so, this approach offers a robust method for controllably seeding multiple cell types.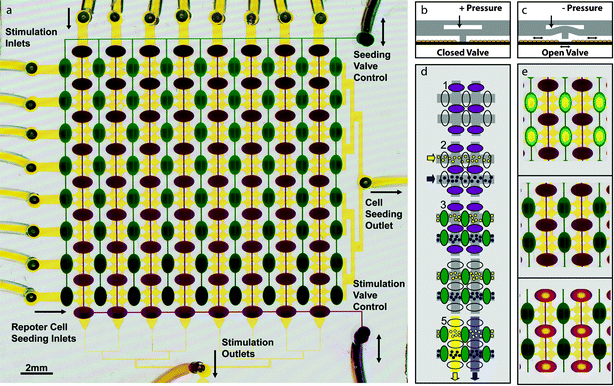 | ||
| Fig. 2 Microfluidic living cell array (a) Layer 1 (yellow) consists of a 16 × 16 array of circular “cell visualization chambers” (50 µm height and 420 µm diameter). Each 2 × 2 subarray in layer 1 is isolated from the others by 2 sets of reversible PDMS barriers. These barriers are controlled by two valve control manifolds (green and purple) in layer 2. Cell lines are drawn from separate inlets (left) through a common outlet (right) to seed the device with rows of different reporters. Similarly, each stimulus is drawn from separate inlets (top) through a common stimulation outlet (bottom). Layer 2 seeding valves (green) are dead-end channels controlled by the pressure in a single inlet (top right) and stimulation valves (purple) are controlled by a single control line (bottom right). (b) Cross-sectional schematic of the reversible barriers. At rest or when positive pressure is applied to the control line, valves are closed. (c) When negative pressure is applied to the control line, the reversible barrier is elevated allowing fluidic communication. (d) Schematic of a typical experiment. 1. Devices are placed in “seeding configuration” with seeding valves open and stimulation valves closed. 2. Reporter cell lines are introduced from the left. 3. The array is placed in neutral configuration by closing seeding valves, and cells are allowed to attach. 4. Devices are then placed in “stimulation configuration” by opening stimulation valves and closing seeding valves. 5. Stimuli are drawn through each column of the array to stimulate each cell line with each stimulus and create a matrix of 64 stimulus–response experiments, each with 4 cell chambers or “replicates”. (e) Images of the dye-filled device in each of the three configurations—seeding (top), neutral (middle), stimulation (bottom). Open valves appear to have yellow centers when the floor and ceiling of the layer 2 control channel meet. Since the dye is squeezed away from that area, the color is dominated by the yellow dye in the underlying layer 1. | ||
To seed cells, arrays are first sterilized, coated with fibronectin, and placed in “seeding configuration” (Fig. 2d–e) by raising seeding valves and leaving stimulation valves closed. Once the array is separated into rows, tubing from each seeding inlet is immersed in a separate concentrated cell suspension (5–10 × 106 cells ml–1) containing a different reporter cell population (see movie in the Electronic Supplementary Information online).† Each reporter cell suspension is manually drawn into each row of the array using a 1 ml syringe (approximately 50 µl of each cell suspension) connected to the common seeding outlet. After seeding, the barriers are returned to their closed positions and cells are allowed to attach and spread inside the array. After 1–2 h, unattached cells are rinsed away and the adherent reporter cells are cultured using either discrete medium changes (2–3 per day) or continuous medium delivery. Discrete medium changes are performed by connecting a medium-filled syringe to the seeding outlet, opening the seeding valves, and advancing the syringe to deliver approximately 100 µl of the medium 2–3 times per day. Alternatively, a continuous flow of medium is delivered at 0.1 µl min–1 using a constant-flow syringe pump. After seeding and attachment (24 h), the microscale cultures are characterized by high viability (∼95% by calcein AM) and active proliferation. Cell density is highly uniform in each row with some variation between rows due to differences in cell suspension preparation. Inside the device, cells assume normal morphologies (Fig. 3a,b) and we routinely maintain cultures for several days until confluency is achieved.
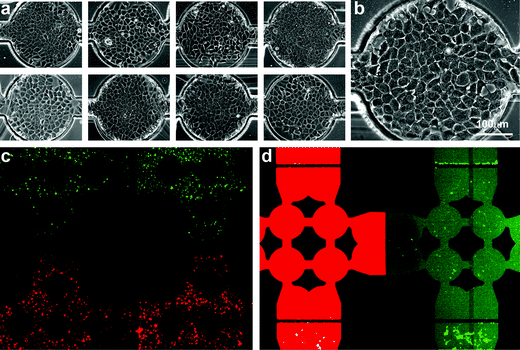 | ||
| Fig. 3 Cell culture in the microfluidic array—(a) Phase contrast images of 8 representative wells in the array. (b) Enlarged phase contrast image of a single confluent cell-visualization chamber with cells exhibiting morphologies similar to those observed on conventional tissue culture plastic. (c) Fluorescence overlay of red- and green-labeled cells seeded in adjacent rows. Valves are closed to separate rows and columns during cell attachment. (d) Fluorescence overlay of calcein red and green being delivered through adjacent columns while the array is in stimulation configuration. | ||
Spatial encoding of multiple cell types is critical for seeding the reporter library in the array. To demonstrate isolation of rows during cell seeding and the prevention of row-to-row communication or “seeding cross-talk,” we labeled cells with red or green cell-tracker and loaded them in alternating rows. These experiments revealed no row-to-row communication (Fig. 3c) and we observed that cells retained their patterns throughout attachment and spreading, and over several days of culture.
To generate a two-dimensional matrix of experiments, the array must be converted from rows to columns to allow perpendicular delivery of soluble stimuli. Once cells are attached and spread, the array is placed in “stimulation configuration” by applying negative pressure to stimulation valves and positive pressure to seeding valves to ensure complete closure. Soluble stimuli are then drawn into the array in parallel using a constant flow syringe pump, either by drawing fluid from 8 stimulus-containing reservoirs into a single outlet syringe (0.8 µl min–1 total flow) or by delivering stimuli from the inlet using a multi-channel syringe pump (0.1 µl min–1 for each of 8 stimulus-containing syringes). To demonstrate isolation of soluble stimuli, we delivered green and red calcein dye through alternating channels. Despite varying inlet pressures to promote cross-talk, the fluorescent solutions remained isolated in columns with no measurable column-to-column communication or “stimulus cross-talk” (Fig. 3e). Taken together, these studies demonstrate that the microfluidic array provides a reliable and scalable method for creating dense two-dimensional arrays of dynamic stimulus–response experiments using living cells.
Reporter library construction and characterization
To monitor real-time gene expression in the microfluidic arrays, we constructed a library of fluorescent protein reporter cell lines representing key pathways involved in inflammation. The reporter cells were constructed by transfecting H35 hepatoma cells with plasmid DNA encoding a fluorescent protein under the transcriptional control of different transcription factors. The result is a cell that expresses the fluorescent protein whenever the transcription factor of interest is active. Specifically, four repeats of transcription factor response elements (Table 1) were cloned upstream of a CMV minimal promoter and a destabilized enhanced green fluorescent protein (d2EGFP) reporter as previously described.18,19 In constructing the reporter library, we aimed to (i) minimize copy number variation across time and enable long-term experimentation by stable transfection, (ii) maximize assay dynamic range by positive and negative fluorescence activated cell sorting (FACS) to select cells with high inducibility and low background in the uninduced state, (iii) minimize population heterogeneity using limiting dilution to obtain monoclonal cell lines, and (iv) allow transient responses to be followed by using a destabilized d2EGFP with a 2 hour half-life as the fluorescent reporter protein. Although spectral variants of GFP could have been used to distinguish different reporter cell lines, we wanted to build a scalable platform that was not limited by the number of available fluorescent protein variants. Therefore, we designed the reporter library using a single d2EGFP reporter, and spatially encoded reporter identities by seeding each cell line in a separate row of the microfluidic array. Each reporter plasmid was transfected into H35 rat hepatoma cells, a cell line chosen for its well-characterized response to inflammatory cytokines. In addition, a constitutively expressing cell line stably transfected with a 4 hour half-life protein (d4EGFP) was used to establish an exposure time that would use the entire dynamic range of the digital camera without saturating the detector. The nontransfected H35 cells (NT) were used as a negative control to detect changes in cellular auto-fluorescence due to variations in morphology during each experiment.| Response element/transcription factora | Consensus sequences | Reporter response element sequences | Inducers | Functions |
|---|---|---|---|---|
| a Stable monoclone reporter cell lines for each transcription factor are referred to by the names in bold. | ||||
| NFκB binding element/NFκB | GGGAMTNYCC26 | GGGAATTTCC | TNF-α | Proinflammatory, Anti-apoptotic |
| AP-1 binding element/AP-1 | TGASTMA26 | TGAGTCA | IL-1 | Proinflammatory, Mitogenic |
| STAT3 binding element/STAT3 | TT(N)4-5 AA27 | TTCCCGAA | IL-6 | Proinflammatory, Anti-apoptotic. |
| ISRE/IRF | SAAA(N)2-3AAASY28 | GAAACTGAAACT | IFN-γ | Proinflammatory |
| GRE/GR | AGAACANNNTGTTCT26 | AGAACAAAATGTTGT | Dexamethasone | Anti-inflammatory |
| HSE/HSF | CNNGAANNTTCNNG29 | CTAGAATGTTCTAG | 42 °C | Cytoprotective |
| CMV-D4EGFP/D4G | — | — | Positive control | — |
| Nontransfected/NT | — | — | Negative control | — |
We programmed an automated microscope to capture fluorescence images from each cell-containing-chamber at regular intervals, and we quantified the dynamic reporter responses using automated image processing and analysis routines written in MATLAB as described in the Methods section. To characterize the approach, we quantified dynamic responses of the Nuclear Factor κB (NFκB) reporter after exposure to Tumor Necrosis Factor-α (TNF-α) (25 ng ml–1), as well as the Glucocorticoid Response Element (GRE) reporter after exposure to Dexamethasone (Dex) (4 µM) using automated microscopy in microfluidic channels as well as the more labor intensive and destructive fluorescence flow cytometry. Comparisons of normalized fluorescence measured by cytometry and microscopy revealed that both techniques captured similar temporal trends and were able to distinguish the distinct dynamics of the two reporters (Fig. 4a–c). Furthermore, because automated time-lapse microscopy allows sampling frequency to be increased freely without requiring additional experiments, we were also able to sample cellular fluorescence at more regular intervals and higher temporal resolution than flow cytometry. In summary, the use of automated time-lapse fluorescence microscopy allows reproducible, high-sensitivity, and high-frequency measurements of cellular fluorescence, making it an ideal method for quantifying reporter dynamics in the microfluidic real-time gene expression array.
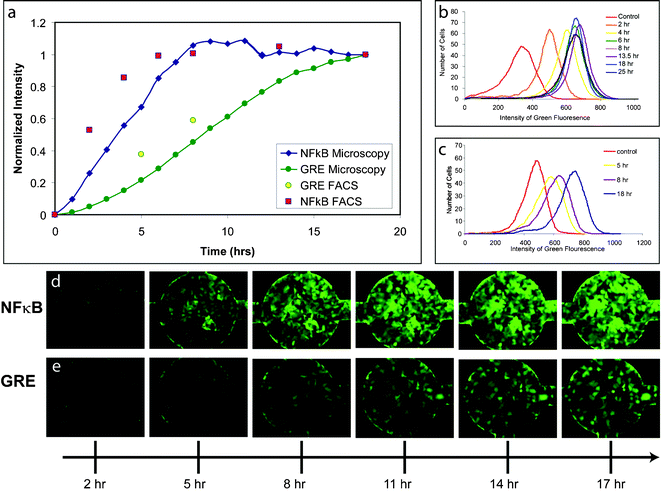 | ||
| Fig. 4 GFP reporter cell line dynamics—(a) Comparison of NFκB and GRE reporter dynamics quantification using FACS and microscopy with image analysis of cells in microfluidic channels. FACS analysis of (b) NFκB and (c) GRE reporter cell populations at 0, 5, 8, and 18 h after stimulation with 25 ng ml–1 TNF-α and 4 µM dexamethasone, respectively. Fluorescence time lapse images of (d) NFκB and (e) GRE reporters in microfluidic cell visualization chambers 2, 5, 8, 11, 14, and 17 h after stimulation. | ||
Dynamics of hepatocyteinflammatory responses
We applied the living cell array to study the hepatocyteinflammatory response by exposing the reporter library (Table 1) to bacterial endotoxin (lipopolysaccharide or LPS), cytokiness, hormones, and their combinations. During infection and inflammation, cells of the innate immune system transiently release large amounts of cytokines, which combine with systemic elevations in stress hormones to elicit a variety of responses in nearby parenchymal cells. Together, the complex soluble inflammatory environment leads to dynamic stimulation of several intracellular signaling pathways, which converge on multiple transcription factors to regulate gene expression changes and coordinate cellular responses. Therefore, we used the microfluidic array to monitor the dynamics of key transcription factors in this response.We seeded the eight cell lines in the microfluidic array, and stimulated the library with various pro- and anti-inflammatory mediators including endotoxin, inflammatory cytokines, a synthetic glucocorticoid, and combinations thereof. Each stimulus was drawn from a separate supply tube through a common outlet using a constant flow syringe pump. Stimulus outlet channels were designed with high resistance to avoid retrograde flow from one stimulus to another. In all experiments, flow rates were chosen to achieve cell surface shear stresses less than 0.1 dynes cm–2. During each experiment, fluorescence dynamics were continuously monitored using time-lapse imaging of each reporter–stimulus pair—192 locations (3 replicates for each of 64 stimulus–reporter pairs) sampled at 90 min intervals. For each reporter–stimulus pair, three of the four replicates were chosen for imaging. This allowed imaging of all locations to be comfortably completed within the 90 min sampling interval and provided us with the flexibility to exclude array elements that were likely to generate artifacts during automated image analysis, such as those containing cell aggregates, acellular debris, or other unintended sources of fluorescence background.
To obtain a global view of dynamic reporter responses to the panel of stimuli, each fluorescence image was quantified and normalized by the maximum and minimum fluorescence for that location as described in the Methods section. The results of a single experiment were then plotted as a heat map (Fig. 5a). Normalizing in this manner not only corrects for the number of cells in each well, but it also highlights temporal aspects of the responses rather than focusing on response magnitudes. One drawback of normalizing data in this manner is that unresponsive stimulus–reporter pairs are rescaled using very low fluorescence levels, which amplifies image noise and results in response dynamics characterized by rapid full-scale fluctuations (see responses to dexamethasone). Nevertheless, as expected, transcriptional reporters in each region of the array exhibited unique fluorescence levels. Negative control cells (NT) consistently exhibited the lowest absolute fluorescence levels while positive control cells (D4G) exhibited the most intense fluorescence for the duration of the experiment, indicating that cell seeding was well controlled. In contrast to NT and D4G responses, the fluorescence levels of the inducible clones were initially low, but increased and decreased in a stimulus-dependent fashion.
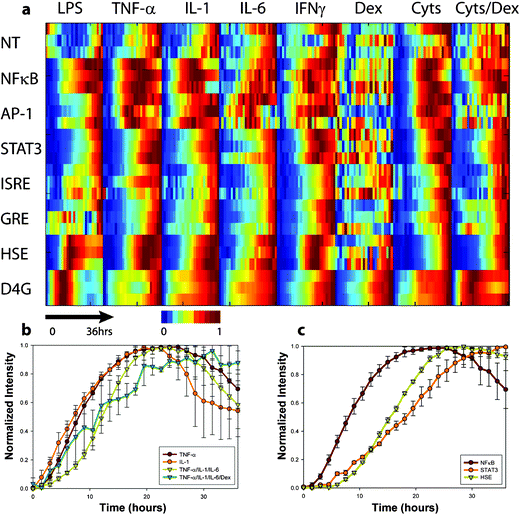 | ||
| Fig. 5 Profiling hepatocyte inflammatory gene expression dynamics (a) Heat map of a single microfluidic living cell array experiment. Each reporter was stimulated with bacterial endotoxin (LPS—25 µg ml–1), inflammatory cytokines(TNF-α—25 ng ml–1, IL-1—25 ng ml–1, IL-6—25 ng ml–1, and IFNγ—10 ng ml–1), a synthetic glucocorticoid hormone (dexamethasone, 4 µM), and combinations thereof (Cyts = TNF-α/IL-1/IL-6) or (Cyts+Dex = TNF-α/IL-1/IL-6/Dex). Cellular fluorescence was measured from 3 cell chambers for each of the 64 stimulus–response pairs every 90 min for 36 h to create the 192 time series comprised of 4608 single-time-point measurements. Data was normalized to initial and maximum levels to highlight the time course of the responses. (b) Responses of NFκB reporters to TNF-α, IL-1, (TNF-α/IL-1/IL-6), and (TNF-α/IL-1/IL-6/Dex). (c) Responses of NFκB, STAT3, and HSE reporters to TNF-α stimulation. | ||
We examined specific stimulus–response interactions using row-by-row analysis of each reporter and compared them to canonical signaling pathways. NFκB was strongly induced by LPS, tumor necrosis factor-α (TNF-α), interleukin 1 (IL-1), interleukin-6 (IL-6), and by combinations of TNF-α, IL-1, and IL-6 (Fig. 5b). Interestingly, while the peak fluorescence varied between these stimuli, the dynamics of the NFκB reporter responses were strikingly stimulus-independent. Indeed, LPS, IL-1, and TNF-α are known to converge on a common signaling pathway involving inhibitor of κB kinase (IKK) phosphorylation and proteosome degradation of the NFκB inhibitor, IκBα.20 Responses were rapid, beginning at 2 h, and reaching a maximum by ∼10 h for each activating stimulus. In contrast, IL-6, a well-characterized protein product of NFκB-mediated transcription, did not cause induction of NFκB. This might be expected, as IL-6 is functionally downstream of NFκB activation in the cytokine signaling cascade. When we added the synthetic glucocorticoid dexamethasone (12 µM) to the cytokine mixture, we observed a significant reduction in NFκB reporter responses, consistent with its established anti-inflammatory properties and its known ability to antagonize NFκB-mediated gene expression. At lower doses of glucocorticoid (4 µM) however, TNF-α regained its ability to elicit a characteristic NFκB response. One surprising result of our experiment was that dexamethasone (Dex) did not elicit a strong GRE reporter response. We suspected that this was an artifact related to using late passage number cells, and after thawing new cells, found that the responsiveness to dexamethasone was restored. Therefore, taken together, our results agree with an extensive body of literature on activation and modulation of NFκB and they serve to validate the microfluidic real-time gene expression platform.
One can gain additional insight by analyzing the array in columns and investigating the coordinated response of multiple transcription factors to a single stimulus. For example, TNF-α resulted in activation of NFκB, Signal Transducer of Activated Transcription 3 (STAT3), and Heat Shock Element (HSE), however the kinetics were distinctly different, with response times (time to 50% of maximum induction) of approximately 5, 12, and 15 h respectively (Fig. 5c). Results such as these illustrate how the real-time gene expression array can paint a dynamic picture of the relative response dynamics of multiple inflammatory transcription factors in a single experiment.
While the majority of our results were in agreement with conventional signaling models, some results were unexpected. For example, when the HSE reporter was exposed to TNF-α, IL-1, or their combinations, we observed increases in fluorescence beginning at ∼8 h. We found only one report of such an effect in the literature,21 and to our knowledge, little is known about the underlying mechanism. At first glance, activation of HSE by TNF-α and IL-1 might suggest an NFκB-mediated mechanism; however we also found that the addition of dexamethasone, while able to attenuate NFκB activity, did not decrease TNF-α-induced activation of HSE. Unexpected effects such as these will be interesting topics for future investigations using more conventional assays. Nevertheless, they demonstrate the utility of the real-time gene expression array for detecting new interactions and instructing the design of additional experiments.
The activation of STAT3 by TNF-α was also unexpected. A possible explanation for this interaction might involve indirect activation through secreted IL-6. TNF-α is known to induce expression of IL-6, and IL-6 is a well-established activator of STAT3. To probe indirect effects that potentially depend on paracrine signaling, one might rearrange the order of the reporters and compare the responses of clones in upstream and downstream positions of the array. Alternatively, putative paracrine communication mechanisms can be studied by adjusting the stimulus flow rate, as this should modulate the balance between convective transport and secretion and thus affect the amount of paracrine signaling.
Discussion
Historically, studies of signal transduction and gene expression have focused on establishing the molecular connections within individual pathways. In an effort to create a more dynamic and integrated picture of the cell, we have developed a microfluidic real-time gene expression array to monitor the coordinated activity of multiple pathways. The platform uses microvalve arrays to achieve reliable seeding and orthogonal stimulation of multiple fluorescent reporter cell lines while enabling automated time-lapse microscopy to continuously monitor dynamic responses from the two-dimensional matrix of experiments. We used the platform to characterize dynamic responses to mediators of the hepatic inflammatory response and in each experiment, we collected 192 time courses consisting of ∼5000 single-time-point measurements (64 reporter–stimulus combinations measured across 25 time points in 3 separate cell visualization chambers or “replicates”). Results are in close agreement with published literature and occasionally, measurements revealed unexpected responses that are guiding formulation and testing of new hypotheses.The microfluidic reporter array offers several advantages over conventional gene expression assays. First, sampling frequency can be freely increased, allowing cell responses to be measured at high temporal resolution and regular intervals. Second, the small channel height reduces fluorescence background associated with culture medium compared to conventional open-volume culture dishes, dramatically improving reporter signal detection. Third, the use of microscopy allows adherent cells to be monitored in their native configurations, retaining links between fluorescence measurements and other cell parameters such as morphology, location, and history, ultimately enabling single cell measurements and facilitating correlations between dynamic responses and endpoint measurements such as cell fate. The microfluidic arrays have significant scaling potential in both the number of cell lines that can be examined and the complexity of stimuli that can be delivered. A natural extension of the current design would involve addition of upstream microfluidic circuits to generate precise and controllable concentrations and combinations of stimuli from a limited number of inputs.
Despite the apparent complexity of the microfluidic arrays, they are remarkably straightforward to construct and operate. Once photolithographic masters are created, arrays can be fabricated, sterilized, and seeded with cells in a single day. Valve control of cell seeding and stimulation requires only two 1 ml syringes regardless of the array size, and continuous flow culture and stimulation can be performed using a single syringe pump. Together, these features make the array broadly accessible to cell biology labs that might not be equipped with specialized fabrication facilities or elaborate fluid control equipment.
Fluorescent protein reporters offer a non-destructive means of functionally characterizing gene regulatory sequences in real-time. In contrast to more proximal and destructive measurements such as DNA binding or mRNA abundance, reporter assays allow continuous measurement of the distal protein product and can distinguish between productive (mRNA producing) and unproductive transcription factor binding events. This is an important distinction for transcription factors such as glucocorticoid receptor, where DNA binding does not necessarily result in productive transcription. The microfluidic reporter array can also be used to study dynamics of specific genes by creating reporters with natural promoters. In summary, the use of fluorescent reporters to study transcriptional regulation provides an opportunity to observe the coordinated dynamics of many genes across time and create an integrated picture of gene expression programs.
Large temporally resolved stimulus–response data sets have previously been assembled using conventional techniques, however substantial investments in time and resources were required. For example, a recent study noted that 18 months were required for 4 investigators to assemble a compendium of stimulus–response dynamics (19 responses to 10 inputs at 10 time points measured in triplicate).22 Nevertheless, the results of these and other dynamic expression studies provide clear evidence that a well-orchestrated temporal order underlies cellular responses in the first 48 h following many experimental stimuli.23
In this work, we demonstrated the microfluidic dynamic gene expression platform using a well-characterized inducible pathway. Once validated however, the technology can be applied more generally to complex gene regulatory networks, such as those found in hepatocytes,24 pancreatic cells,24 and embryonic stem cells,25 where transcription factors are themselves, transcriptionally regulated. The microfluidic real-time gene expression array provides a powerful means of collecting large amounts of quantitative dynamic data from living cells in an internally consistent format, aiding development of mathematical models of gene regulatory networks and high-throughput investigations of signaling dynamics for systems biology. Furthermore, the unbiased nature of these experiments creates an opportunity to broadly characterize complex signaling networks and discover “off-target” effects of putative therapeutics. In the future, profiling tools such as the microfluidic real-time gene expression array have the potential to play an important role in revealing the systems dynamics of the cell, uncovering unexpected mechanisms of drug actions, and developing clinical fingerprints of disease.
Methods
Microfluidic array fabrication
Microfluidic arrays are fabricated by replica molding PDMS from photolithographically defined SU-8 masters. Layer 1 molds are spin-coated with PDMS (1500 rpm for 15 s) and Layer 2 is cast to a thickness of 2–5 mm. To assemble the array, valve control inlets are drilled with a blunt and beveled needle and the Layer 2 PDMS is manually aligned and bonded to the thin silicon-supported Layer 1 PDMS assisted by oxygen plasma surface treatment and heating (100 °C, 2 h). The Layer 2 photomask was printed at 101.8% of the target dimensions to correct for contraction of the PDMS after release from the silicon master. After bonding, the Layer 1–Layer 2 stack is removed from the silicon support, inlets are drilled for connection to Layer 1 and the PDMS is bonded to a glass slide again using oxygen plasma and heating. To avoid permanent bonding of valves to the underlying glass, valves are held open by applying negative pressure to layer 2 control lines during bonding to glass, by patterning metal bond pads on the glass at the location of the valves as previously described,16 or by blocking valve surfaces during oxygen plasma treatment using a PDMS stamp. Once bonding is complete, devices are filled with liquid and valves are repeatedly cycled to prevent subsequent sticking.Reporter cell line construction and characterization
Reporter plasmidswere designed to have multiple response elements (sequences that bind the transcription factor of interest), upstream of a minimal promoter (to prevent high background expression in the absence of the transcription factor of interest), upstream of a destabilized fluorescent protein gene (to encode the d2EGFP reporter protein). The reporter plasmid was developed by cloning multiple copies of response element (RE) repeats into pCMVmin-EGFP-1, which was constructed using pEGFP-1 (Clontech) with an insertion of a CMV minimal promoter between Kpn I and Sma I digested from pTRE-d2EGFP (Clontech). REs were inserted before CMVmin between Bgl II and Hind III on pCMVmin-EGFP-1. The EGFP gene was replaced with that of d2EGFP from pd2EGFP-1 (Clontech) using BamH I and Not I. The 2 h EGFP variant was chosen to enable continuous monitoring of dynamic responses because it has a short half-life and it does not accumulate indefinitely. DNA transfection of H35 cells was performed by electroporation, and G418 (0.7 mg ml–1) selection antibiotic was used to isolate stably transfected cells. Using flow cytometry, stably transfected cells were sorted for positive responses to classic inducers (Table 1) and negative responses in the absence of inducers to identify cells with high inducibility and low baseline expression. Limited dilution and further FACS selection was performed to obtain several monoclonal reporter cell lines with high dynamic range. Clone characterization was performed by measuring fluorescence dynamics in response to classic inducers at discrete time points by FACS.Microfluidic seeding, culture, and stimulation
Devices were UV sterilized, coated with fibronectin (25 µg ml–1) for at least 2 h at 37 °C and degassed by driving trapped air through the gas-permeable device walls with positive pressure. Dead-end valve control lines were filled with phosphate buffered saline (PBS), and culture chambers were seeded with cells as described in the text. Cells were maintained in culture using discrete medium changes twice daily. Discrete medium changes were performed by opening the appropriate valves and advancing a medium-containing syringe. Stimulation experiments were performed using a customized incubated microscope stage at 37 °C in phenol red-free serum-free DMEM containing 25 mM HEPES buffer. Calcein and cell tracker dyes were obtained from Molecular Probes. Stimuli were delivered to cells from 1 ml syringes controlled by a multichannel syringe pump (Harvard Apparatus, Holliston, MA). TNF-α, IL-1, and IL-6 were obtained from R&D Systems and used at 10–25 ng ml–1. LPS and dexamethasone were obtained from Sigma and used at 25 µg ml–1 and 4–12 µM respectively. Constant flow rate stimulations were performed at 0.1 µl min–1. Images were captured in phase and fluorescence at pre-programmed locations every 90 min for the duration of the experiment (36 h). Devices were monitored using automated time-lapse fluorescence microscopy and quantified using automated image analysis.Image analysis and quantification
Fluorescence images were captured on a Zeiss 200 Axiovert microscope using an AxioCAM MRm digital camera and quantified using custom image analysis routines written in MATLAB (Mathworks, Natick, MA). Each image was divided by an image with uniform fluorescence to correct for spatial variations in fluorescence excitation. The intensity histogram of each image was then combined with a user-defined “threshold parameter” to automatically determine and subtract a background fluorescence level. For example, the data shown in Fig. 5 was processed with a threshold parameter of 0.2, which identified the fluorescence level below which 20% of pixels resided, interpreted it to be the background level, and subtracted it from the entire image. This method for identifying background fluorescence is justified because ∼20% of each image consisted of PDMS without cells. The subtraction is justified by assuming that cells have a small thickness compared to the culture medium and PDMS. Therefore, the cells do not significantly displace other sources of background and are assumed to be additive with background fluorescence. Because this procedure was performed at each time point, it corrected for temporal variations in excitation source intensity. The fluorescence of the post-processed image was integrated to generate a single measurement for each cell-containing well. Measurements were organized by location to create a fluorescence response time series for each element of the microfluidic array and all measurements were assigned to the imaging time of the first array element. To highlight temporal aspects of the responses and facilitate comparison between locations, each response was normalized between its maximum and minimum fluorescence levels according to Φij(t) = [Fij(t) – Fij_min]/[Fij_max – Fij_min] where Φij(t) is the normalized fluorescence of row i and column j at time t, Fij(t) is the post-processing image fluorescence, and Fij_max and Fij_min are the maximum and minimum post-processing fluorescence values for the ij array location respectively. Normalizing in this manner makes responses independent of the number of cells in each array element, however it amplifies noise related to automated imaging and analysis and results in rapid full-scale fluctuations for wells in which stimulus-reporter combinations do not generate robust responses (see responses to dexamethasone in Fig. 5a). In some cases unresponsive cells exhibit morphological changes that lead to more gradual changes in background fluorescence (see NT cell responses) and generate what appear to be meaningful signals, despite the lack of a GFP-related response. We are actively developing image analysis techniques that should significantly improve the quality of the data collected in this platform.Acknowledgements
We would like to acknowledge NIH Grants GM065474 and AI063795, NIH BioMEMS Resource Center Grant P41 EB-002503, and a grant from the Shriners Hospital. The authors would like to thank Pohun Chris Chen, Cindy Zia, Ken Wieder, and Octavio Hurtado for technical support.References
- R. A. Wagner, R. Tabibiazar, A. Liao and T. Quertermous, Dev. Biol., 2005, 288, 595–611 CrossRef CAS.
- V. R. Iyer, M. B. Eisen, D. T. Ross, G. Schuler, T. Moore, J. C. Lee, J. M. Trent, L. M. Staudt, J. Hudson, Jr., M. S. Boguski, D. Lashkari, D. Shalon, D. Botstein and P. O. Brown, Science, 1999, 283, 83–7 CrossRef CAS.
- J. I. Murray, M. L. Whitfield, N. D. Trinklein, R. M. Myers, P. O. Brown and D. Botstein, Mol. Biol. Cell, 2004, 15, 2361–74 CrossRef CAS.
- S. E. Calvano, W. Xiao, D. R. Richards, R. M. Felciano, H. V. Baker, R. J. Cho, R. O. Chen, B. H. Brownstein, J. P. Cobb, S. K. Tschoeke, C. Miller-Graziano, L. L. Moldawer, M. N. Mindrinos, R. W. Davis, R. G. Tompkins and S. F. Lowry, Nature, 2005, 437, 1032–7 CrossRef CAS.
- G. A. Boorman, P. E. Blackshear, J. S. Parker, E. K. Lobenhofer, D. E. Malarkey, M. K. Vallant, D. K. Gerken and R. D. Irwin, Toxicol. Sci., 2005, 86, 185–93 CrossRef CAS.
- A. Zaslaver, A. E. Mayo, R. Rosenberg, P. Bashkin, H. Sberro, M. Tsalyuk, M. G. Surette and U. Alon, Nat. Genet., 2004, 36, 486–91 CrossRef CAS.
- S. Kalir and U. Alon, Cell, 2004, 117, 713–20 CrossRef CAS.
- J. Liu, C. Hansen and S. R. Quake, Anal. Chem., 2003, 75, 4718–23 CrossRef CAS.
- E. M. Lucchetta, J. H. Lee, L. A. Fu, N. H. Patel and R. F. Ismagilov, Nature, 2005, 434, 1134–8 CrossRef CAS.
- B. G. Chung, L. A. Flanagan, S. W. Rhee, P. H. Schwartz, A. P. Lee, E. S. Monuki and N. L. Jeon, Lab Chip, 2005, 5, 401–6 RSC.
- J. L. Tan, J. Tien, D. M. Pirone, D. S. Gray, K. Bhadriraju and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1484–9 CrossRef CAS.
- N. Li Jeon, H. Baskaran, S. K. Dertinger, G. M. Whitesides, L. Van de Water and M. Toner, Nat. Biotechnol., 2002, 20, 826–30 CAS.
- A. Bader, I. H. Borel Rinkes, E. I. Closs, C. M. Ryan, M. Toner, J. M. Cunningham, R. G. Tompkins and M. L. Yarmush, Biotechnol. Prog., 1992, 8, 219–25 CrossRef CAS.
- Y. H. Kang, F. Berthiaume and M. L. Yarmush, Tissue Eng., 2002, 8, 681–93 CrossRef CAS.
- A. Jayaraman, M. L. Yarmush and C. M. Roth, Tissue Eng., 2005, 11, 50–63 CrossRef CAS.
- D. Irimia and M. Toner, Lab Chip, 2006, 6, 345–52 RSC.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–4 CrossRef CAS.
- D. M. Thompson, K. R. King, K. J. Wieder, M. Toner, M. L. Yarmush and A. Jayaraman, Anal. Chem., 2004, 76, 4098–103 CrossRef CAS.
- K. J. Wieder, K. R. King, D. M. Thompson, C. Zia, M. L. Yarmush and A. Jayaraman, Biomed. Microdevices, 2005, 7, 213–22 CrossRef.
- S. L. Werner, D. Barken and A. Hoffmann, Science, 2005, 309, 1857–61 CrossRef CAS.
- G. Schett, K. Redlich, Q. Xu, P. Bizan, M. Groger, M. Tohidast-Akrad, H. Kiener, J. Smolen and G. Steiner, J. Clin. Invest., 1998, 102, 302–11 CrossRef CAS.
- K. A. Janes, J. R. Kelly, S. Gaudet, J. G. Albeck, P. K. Sorger and D. A. Lauffenburger, J. Comput. Biol., 2004, 11, 544–61 CrossRef CAS.
- K. A. Janes, J. G. Albeck, S. Gaudet, P. K. Sorger, D. A. Lauffenburger and M. B. Yaffe, Science, 2005, 310, 1646–53 CrossRef CAS.
- D. T. Odom, N. Zizlsperger, D. B. Gordon, G. W. Bell, N. J. Rinaldi, H. L. Murray, T. L. Volkert, J. Schreiber, P. A. Rolfe, D. K. Gifford, E. Fraenkel, G. I. Bell and R. A. Young, Science, 2004, 303, 1378–81 CrossRef CAS.
- L. A. Boyer, T. I. Lee, M. F. Cole, S. E. Johnstone, S. S. Levine, J. P. Zucker, M. G. Guenther, R. M. Kumar, H. L. Murray, R. G. Jenner, D. K. Gifford, D. A. Melton, R. Jaenisch and R. A. Young, Cell, 2005, 122, 947–56 CrossRef CAS.
- http://www.gene-regulation.com/pub/databases.html#transfac, TRANSFAC database, accessed 2005.
- W. D. Morgan, G. T. Williams, R. I. Morimoto, J. Greene, R. E. Kingston and R. Tjian, Mol. Cell. Biol., 1987, 7, 1129–1138 CAS.
- H. M. Seidel, L. H. Milocco, P. Lamb, J. E. Darnell Jr, R. B. Stein and J. Rosen, Proc. Natl Acad. Sci. U. S. A., 1995, 92, 3041–3045 CrossRef CAS.
- N. Tanaka, T. Kawakami and T. Taniguchi, Mol. Cell. Biol., 1993, 13, 4531–4538 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Cell seeding movie. See DOI: 10.1039/b612516f |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2007 |