Electrophoretic partitioning of proteins in two-phase microflows†
G.
Münchow
*a,
S.
Hardt
b,
J. P.
Kutter
c and
K. S.
Drese
a
aInstitut für Mikrotechnik Mainz GmbH, Carl-Zeiss-Straße 18-20, D-55129, Mainz, Germany. E-mail: muenchow@imm-mainz.de
bDarmstadt University of Technology, Petersenstraße 30, D-64287, Darmstadt, Germany
cTechnical University of Denmark (DTU), Oersteds Plads, Building 345 east, DK-2800, Kongens Lyngby, Denmark
First published on 27th September 2006
Abstract
This work reports on protein transport phenomena discovered in partitioning experiments with a novel setup for continuous-flow two-phase electrophoresis consisting of a microchannel in which a phase boundary is formed in flow direction. Proteins can be partitioned exploiting their affinity to different aqueous phases in two-phase systems. This separation process may be enhanced or extended by applying an electric field perpendicular to the phase boundary. In this context, microsystems offer new possibilities, as interfacial forces usually dominate over volume forces, thus allowing a superior control of the formation and arrangement of liquid/liquid phase boundaries. The two immiscible phases which are injected separately into the microchannel are taken from a polyethylene glycol (PEG)–dextran system. The side walls of the channel are partially made of gel material which serves as an ion conductor and decouples the channel from the electrodes, thus preventing bubble generation inside the separation channel. The experiments show that the electrophoretic transport of proteins between the laminated liquid phases is characterized by a strong asymmetry. When bovine serum albumin (BSA) is introduced into the PEG-rich phase, it can easily be transferred into the dextran-rich phase via an applied electric field of low strength or just by diffusion. In the reverse case, up to a certain field strength the transfer to the opposing phase is strongly inhibited. Only if the field strength is further increased will the BSA molecules leave the dextran-rich phase almost completely.
Introduction
Aqueous two-phase systems (ATPSs) are widely used for the purification and isolation of proteins. These systems are formed by dissolving small amounts of two incompatible polymers, such as polyethylene glycol (PEG) and dextran, or one polymer and a salt above a given concentration. Since both of the resulting phases consist largely of water (80–90% (w/w)), biocompatibility is ensured. Protein partitioning in such a system takes place due to different affinities of a protein species to the polymers.1–4 The affinities and by that the migration across the phase boundary depend on the physico-chemical properties of the protein such as isoelectric point, surface hydrophobicity, molecular mass as well as on medium properties like polymer molecular mass, pH, and salt concentrations.4–8 By controlling these factors, the selective partitioning and recovery of a target protein can be achieved. Corresponding ATPSs have also been used for the partitioning of nucleic acids and cells9–11 and first steps towards a microfluidic implementation of such schemes have been taken.12–14The first attempts at protein partitioning in ATPS supported by an additional electric field have been made by different groups.15–20 Clark et al. have studied the two-phase electrophoretic separation of proteins inside a self-build macro-device. Levine and Bier have examined the partitioning coefficients and electrophoretic mobilities of proteins inside a U-tube. Both groups noticed some accumulation of proteins at the phase boundary when using specific buffer types.
In the present study, bi- or multilaminated ATPSs based on PEG and dextran are formed in a microchannel to examine the electrophoretic transport of proteins towards and across the phase boundary. Due to the fact that both phases are electrically conductive, an electric field perpendicular to the phase boundary can be applied enabling transport not only from the phase with the lower partitioning coefficient to the preferred phase but also vice versa.
By means of this work a basis for novel microfluidic two-phase systems combined with electrophoretic separation should be established. Potential applications lie in the field of purification and separation of biomolecules for which the combination of electrophoresis and two-phase partitioning should offer new possibilities.
Materials and methods
Materials
Bovine serum albumin (BSA, Fluka-05480), dextran of 500 000 Dalton (Da) average molecular weight (range: not specified, D 5251) and PEG of 8000 Da average molecular weight (range: 7000–9000 Da, P 2139) were obtained from Sigma-Aldrich (Germany). Water was deionized and further purified using a Millipore Milli-Q water treatment unit. The BSA molecules were labelled with fluorescence markers using the Alexa Fluor® 488 Protein Labeling Kit (A-10235, Molecular Probes/Invitrogen, Germany).The experiments were performed with 60 µg ml−1 of labelled BSA dissolved in either the PEG or the dextran phase. The two-phase system was prepared by dissolving 3.8 wt% PEG and 5.5 wt% dextran in a buffer solution (potassium phosphate, 5 mM) at different pH values (pH 6, pH 7 and pH 8). After stirring and settling overnight the phases separate with a sharp stable phase boundary between them. The upper PEG-rich phase (PEG-phase) consists of 5.7 wt% PEG, 1.0 wt% dextran and 88.6 wt% of buffer solution. The lower dextran-rich phase (Dex-phase) consists of 1.9 wt% PEG, 9.5 wt% dextran and 93.3 wt% of buffer.15 Finally, the two phases were separated by a pipette and stored in different vessels. The gel enabling a fluidic decoupling of the microchannel from the buffer reservoirs consists of 1.5 wt% agarose (Sigma, Germany) and is based on the same buffer as used for preparing the PEG/dextran two-phase system.
Microdevice fabrication and design
The polymethylmethacrylate (PMMA) microdevice is fabricated using a CNC micro milling machine and is sealed temporarily by adhesive tape or permanently by attaching a PMMA film using a solvent bonding process. The microchannel design is shown in Fig. 1. The chip consists of three inlets and one outlet. The width, depth and length of the separation channel are 800 µm, 120 µm and 30 mm, respectively. It is located between comb structures which are filled by agarose gel separating it from the buffer reservoirs on the backside of the chip where electrolysis takes place. The gel matrix enables a fluidic decoupling and serves as an ion conductor through which the electric field is guided. The channel comprises numerous gel bridges on each side (the “teeth” of the comb structure) each having a cross section of 350 µm × 100 µm.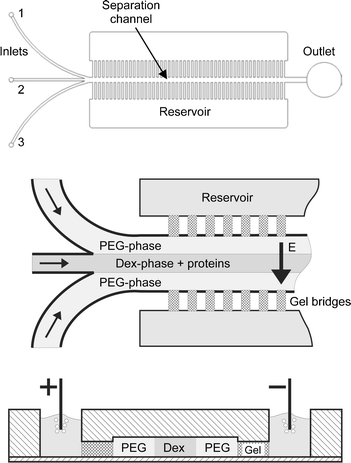 | ||
| Fig. 1 Upper: CAD drawing of the microdevice. The microfluidic chip comprises a separation channel with three inlets and one outlet. Middle: Injection procedure. Lower: Cut perpendicular through the microchannel showing the reservoirs and the separation channel in the center. | ||
After the channel structure is sealed the agarose gel solution is heated up to 100 °C and filled into the comb structure from the backside of the chip. Driven by capillary forces the gel solution flows towards the separation channel and stops at the openings to the channel which act as barriers for hydrophilic solutions. The comb structures are connected to reservoirs on the backside of the chip which, after the agarose solution has gelled, are filled with the same buffer as used for preparing the two-phase system, cf.Fig. 1.
Experimental procedure
The PEG- and Dex-phases are co-injected into the microfluidic structure using syringe pumps (KD Scientific Inc., MA, USA) at different flow rates. The BSA molecules are dissolved in the Dex-phase which is injected through inlet 2, cf.Fig. 1. This phase is embraced by the PEG-phase fed to inlets 1 and 3 such that a system of three co-flowing liquid lamellae is formed. Due to the different viscosities of the two phases the flow velocities have to be adjusted in the lamellae of different types to achieve uniform lamella widths. For the experiments reported in this article, the flow rates were chosen to be 0.1 ml h−1 and 0.03 ml h−1 for the PEG- and Dex-phase, respectively. The corresponding residence time within the electric field for BSA molecules dissolved in the Dex-phase is approximately 6 minutes and long enough to influence their distribution across the channel significantly. The protein distribution is recorded by mapping the fluorescence intensity using a fluorescence microscope (CKX 41, Olympus, Germany) with a CCD camera attached to it (Kappa DX 30 C, Germany).Results
Hydrodynamic regimes and stability of laminated flow
Two of the most important parameters governing the electrohydrodynamic response of the laminated two-phase system, sketched in Fig. 1, are the interfacial tension and the electric permittivity of the two liquid phases. Systems with a very large interfacial tension, like oil–water, tend to become unstable when bilaminated into a channel,21 whereas systems with a small interfacial tension usually have less stability problems, but consist of components which are more similar physico-chemically. For the two immiscible phases of aqueous solutions of PEG and dextran the dielectric permittivities are similar and the interfacial tension is quite small (0.1–0.001 mN m−1).22,23A surface-tension driven instability of two-phases co-flowing in a microchannel usually leads to droplet formation.21 However, the occurrence of such an instability depends on the aspect ratio (height/width) of the liquid lamellae. Choosing a small aspect ratio minimizes the influence of the liquid/liquid phase boundary and stabilizes the flow. A stable laminated flow of PEG- and Dex-phases was observed for aspect ratios up to approx. 0.2.
The flow pattern in the multilaminated two-phase flow is also influenced by the electric field. It was found that at high applied voltages perturbations develop on the phase boundary, cf.Fig. 2. These perturbations are caused by an inhomogeneous distribution of ions and have been exploited for example in mixing in microchannels.24,25 However, below a critical voltage of about 15 V dc, depending on the salt concentration of the buffer solution (here 5 mM), the laminated flow remains stable.
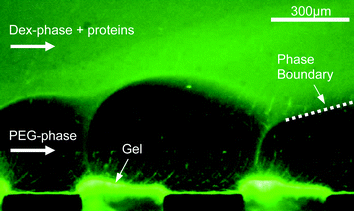 | ||
| Fig. 2 Perturbations of the phase boundary in high electric fields. Only the lower PEG phase and two of the gel bridges are visible. Proteins already accumulate at the gel surface. | ||
Transport of BSA towards the phase boundary
In the first part of the test series fluorescence labelled BSA molecules are dissolved in the Dex-phase, as shown in Fig. 1. If the system is based on a potassium phosphate buffer at low pH values, the BSA molecules prefer the Dex-phase while transport to the PEG-phase is largely suppressed. By contrast, if BSA molecules are first dissolved in the PEG-phase they immediately diffuse across the phase boundary into the preferred Dex-phase without any observed hindrance. These phenomena indicate a pronounced asymmetry of protein transport between the two phases. Possibly the free energy barrier derived from protein partition coefficients and electric double layers at the phase boundary play a role in this context. Typically, the partition coefficient for BSA varies from 0.01826,27 to 0.629 depending on buffer type and polymer concentration.For each experimental run using the microdevice, a known amount of BSA is dissolved in the preferred Dex-phase yielding a concentration of 60 µg ml−1. The two different phases are injected into the microchannel, creating a three-lamella arrangement, cf.Fig. 1. After ten minutes of equilibration time allowing the system to become stable and to form equal lamella widths, the first image is recorded with the CCD camera, where the electric field is switched off. The equilibration time is needed because first the channel is completely filled with the PEG-phase and afterwards the dextran lamella is injected. Owing to the small flow velocities in the experiments, it takes some time until the final three-lamella arrangement is fully developed over the complete channel length. After that an electric field is applied and increased stepwise. The next images are recorded five minutes after each increase of the electric field strength, giving the system enough time to respond to the change in electric field. All images are taken at a position 30 mm downstream of the confluence of the three feed streams close to the exit of the channel.
Up to approximately 2.5 V dc the majority of BSA molecules are confined within the Dex-phase but move towards one of the phase boundaries to the neighboring PEG-phase, depending on the applied electric field direction. This leads to a non-uniform fluorescence intensity distribution across the dextran lamella and a concentration at the phase boundary, cf.Fig. 3. The bell-shaped curve obtained at 0 V dc with a fall-of towards the fluid interfaces can be explained by assuming a slow diffusive mass transfer to the neighboring phase. Thus, the initially rectangular concentration profile is smeared out by diffusion, which is so slow that it does not lead to a significant depletion of the Dex-phase within the time scales considered here.
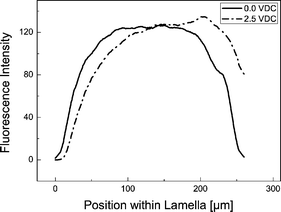 | ||
| Fig. 3 Fluorescence intensity across the dextran-rich lamella. In order to smooth out noise, an average over 15 neighboring data points was taken. The phase boundaries to the PEG-rich lamellae are located at x-values 0 and 220 µm, respectively. At 0.0 V dc the fluorescence shows a uniform distribution with a fall-off towards the phase boundaries at the left and at the right. At 2.5 V dc the BSA molecules move towards one phase boundary which leads to a displacement of the intensity curve depending on the electric field direction. Buffer: potassium phosphate, 5 mM, pH 7.0. | ||
The concentration of proteins at the phase boundary was also discussed by Levine et al.18 and Theos et al.17 They have found evidence of such an accumulation in systems with phosphate buffer, known to provide a significant electric double-layer potential (1.5 to 2.2 mV, depending on the pH value).28,29 An interfacial accumulation was not observed for example, in Tris/Bes buffer with a low double-layer potential of 0.3 mV.17 Due to the macroscopic setup of Levine et al. and Theos et al. a quantitative study of this phenomenon was difficult. The use of a microfluidic setup and fluorescence labelled proteins enables a detailed examination of the influence of the phase boundary in ATPSs on the transport behavior of proteins.
Transport across the phase boundary
If BSA is dissolved in the preferred dextran-rich lamella, up to a specific value the applied electric field mainly leads to a redistribution of proteins, as described in the previous section. After a further increase of the field strength by applying a voltage of more than 2.5 V dc the proteins increasingly overcome the phase boundary, cf.Fig. 4 and 5. Fig. 5 shows the average fluorescence intensity (averaged over the width of the dextran lamella) at the observation point close to the exit of the channel as a function of applied voltage. The data points recorded in the experiments are displayed together with fitting functions of the form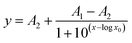 (lines) where x represents the applied voltage. At small voltages a plateau is formed showing that fewer proteins are able to penetrate the phase boundary. When the voltage is increased, the average fluorescence intensity in the Dex-phase shows a steep decrease and finally levels off at a value close to zero, indicating that virtually all of the molecules have been transferred to the PEG-phase. It should be emphasized that the slope in Fig. 5 indicating a decreasing fluorescence intensity does not necessarily reflect the equilibrium of the system, but depends on the kinetics and speed of the transition of proteins between the two phases. Owing to the finite residence time in the microchannel, only the fraction of proteins remaining in the Dex-phase after a specific time span are recorded. In fact measurements conducted at reduced flow rates indicate that the decrease of fluorescence intensity shown in Fig. 5 occurs at a slightly lower voltage. This suggests that the data recorded do not reflect an equilibrium situation, but a dynamic process which shows a nonlinear behavior as a function of the applied voltage.
(lines) where x represents the applied voltage. At small voltages a plateau is formed showing that fewer proteins are able to penetrate the phase boundary. When the voltage is increased, the average fluorescence intensity in the Dex-phase shows a steep decrease and finally levels off at a value close to zero, indicating that virtually all of the molecules have been transferred to the PEG-phase. It should be emphasized that the slope in Fig. 5 indicating a decreasing fluorescence intensity does not necessarily reflect the equilibrium of the system, but depends on the kinetics and speed of the transition of proteins between the two phases. Owing to the finite residence time in the microchannel, only the fraction of proteins remaining in the Dex-phase after a specific time span are recorded. In fact measurements conducted at reduced flow rates indicate that the decrease of fluorescence intensity shown in Fig. 5 occurs at a slightly lower voltage. This suggests that the data recorded do not reflect an equilibrium situation, but a dynamic process which shows a nonlinear behavior as a function of the applied voltage.
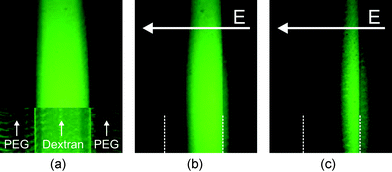 | ||
| Fig. 4 (a) Uniform distribution of BSA within the Dex-phase without electric field; upper part: fluorescence intensity, lower part: fluorescence intensity superimposed on transmitted-light image showing the phase boundaries. (b)–(c) Proteins concentrate at the right phase boundary at 3.5 and 4.5 V dc. At 3.5 V dc a small number of the BSA molecules have already overcome the phase boundary. At 4.5 V dc most of the BSA has left the preferred Dex-phase. Inhomogeneous lighting of the fluorescence light source leads to a reduction of fluorescence intensity at the top of the picture. Buffer: potassium phosphate, 5 mM, pH 7.0. | ||
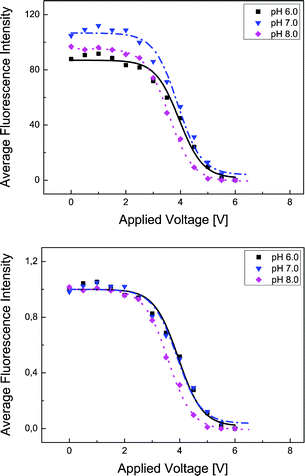 | ||
| Fig. 5 Upper: Average fluorescence intensity within the dextran-rich lamella. The intensities of all pixels within the lamella were added and divided by the number of pixels to eliminate influences from variations in the lamella width. Lower: Normalized intensity curves with all data divided by their values at U = 0 V dc. After a voltage of about 2.5 V dc is reached, the BSA molecules start to penetrate the phase boundary. | ||
The experiments have been performed at three different pH values, pH 6, pH 7 and pH 8. The isoelectric point of BSA lies at a value of approximately pH 4.7, thus showing that the pH variations leave the sign of the protein charge invariant. Rather than studying the influence on the protein charge the pH variations were done to examine a possible influence on the transport barrier between the phases, since it is known that the double-layer potential at the phase boundary is a function of pH.28,29 When normalizing the intensity curves as done in the lower part of Fig. 5, apparent differences between different pH values disappear. The conclusion has to be drawn that the electrophoretic transport of BSA across the phase boundary is largely independent of pH, at least in the range between pH 6 and pH 8. Deriving a pH-dependence from the curves in Fig. 5 seems to be without support, since the gel preparation process in each experiment may lead to slight differences in size of the gel barriers and by that to different electric field strengths within the separation channel at the same applied voltage. Due to this fact, even when fixing the overall voltage to a specific value the voltage drop across the channel may vary slightly.
Conclusions and outlook
In an ATPS based on PEG/Dextran and a potassium phosphate buffer the electrophoretic transport of BSA shows a strongly asymmetric behavior. Proteins initially in the PEG-phase penetrate the phase boundary without any hindrance. If BSA is dissolved in the Dex-phase and an electric field is applied perpendicular to the liquid lamellae, the transport across the phase boundary is negligible up to a certain field strength. In that case, BSA molecules are set into electrophoretic motion, which does not lead to a significant depletion of the phase up to a voltage of about 2.5 V dc. Only after a further increase of the applied voltage do the proteins increasingly leave the preferred phase and the average fluorescence intensity within the lamella starts to decrease. This transport process seems to be independent of the buffer pH value in a range between pH 6 and pH 8.After demonstrating this mass-transfer asymmetry it seems to be worthwhile to study whether the observed transport phenomena can be utilized to separate different types of proteins. Corresponding microfluidic systems could offer a high potential for such applications as purification, isolation and concentration of proteins or more macroscopic objects such as cells or subcellular fragments.
Acknowledgements
This work is supported by the German Research Foundation, priority program 1164.References
- P.-Å. Albertsson, Nature, 1958, 4637, 709 CrossRef.
- P.-Å. Albertsson, S. Sasakawa and H. Walter, Nature, 1970, 228, 1329 CAS.
- V. P. Shanbhag, Biochim. Biophys. Acta 320, 1973, 320, 517 Search PubMed.
- N. L. Abbott, D. Blankschtein and T. A. Hatton, Bioseparation, 1990, 1, 191 CAS.
- G. Tubio, B. Nerli and G. Picó, J. Chromatogr., B: Biomed. Appl., 2004, 799, 293 CrossRef CAS.
- H.-O. Johansson, G. Karlström, F. Tjerneld and C. A. Haynes, J. Chromatogr., B: Biomed. Appl., 1998, 771, 3.
- J. N. Baskir, T. A. Hatton and U. W. Suter, Biotechnol. Bioeng., 1989, 34, 541 CrossRef CAS.
- A. D. Diamond and J. T. Hsu, AIChE J., 1990, 36(7), 1017 CrossRef CAS.
- P.-Å. Albertsson, Partition of Cell Particles and Macromolecules, Wiley and Sons Publishers, New York, 1986 Search PubMed.
- S. L. Zhai, G. S. Luo and J. G. Liu, Sep. Purif. Technol., 2001, 21, 197 CrossRef CAS.
- S. L. Zhai, G. S. Luo and J. G. Liu, Chem. Eng. J., 2001, 83, 55 CrossRef CAS.
- M. Yamada, V. Kasim, M. Nakashima, J. Edahiro and M. Seki, Biotechnol. Bioeng., 2004, 88, 489 CrossRef CAS.
- K.-H. Nam, W.-J. Chang, H. Hong, S.-M. Lim, D.-I. Kim and Y.-M. Koo, Biomed. Microdevices, 2005, 7(3), 189 CrossRef.
- S. Santesson, I. Barinaga-Rementeria Ramírez, P. Viberg, B. Jergil and S. Nilsson, Anal. Chem., 2004, 76, 303 CrossRef CAS.
- W. M. Clark, Chemtech, 1992, 22, 425 CAS.
- M. A. Marando and W. M. Clark, Sep. Sci. Technol., 1993, 28, 425.
- C. W. Theos and W. M. Clark, Appl. Biochem. Biotechnol., 1995, 54, 143 CrossRef CAS.
- M. L. Levine and M. Bier, Electrophoresis, 1990, 11, 605 CrossRef CAS.
- M. L. Levine, H. Cabezas, Jr. and M. Bier, J. Chromatogr., 1992, 607, 113 CrossRef CAS.
- J. Stichelmair, J. Schmidt and R. Proplesch, Chem. Eng. Sci., 1992, 47, 3015 CrossRef.
- H. Pennemann, S. Hardt, V. Hessel, P. Löb and F. Weise, Chem. Eng. Technol., 2005, 28, 501 CrossRef CAS.
- J. Rydèn and P.-Å. Albertsson, J. Colloid Interface Sci., 1971, 37(1), 219 CrossRef CAS.
- Y.-T. Wu and Z.-Q. Zhu, Chem. Eng. Sci., 1999, 54, 433 CrossRef CAS.
- A. O. E. Moctar, N. Aubry and J. Batton, Lab Chip, 2003, 2, 273 RSC.
- N. S. Sundaram and D. K. Tafti, Anal. Chem., 2004, 76, 3785 CrossRef CAS.
- U. Gündüz and K. Korkmaz, J. Chromatogr., B: Biomed. Appl., 2000, 743, 255 CrossRef CAS.
- L. Fele and M. Fermeglia, J. Chem. Eng. Data, 1996, 41, 331 CrossRef CAS.
- D. E. Brooks, K. A. Sharp, S. Bamberger, C. H. Tamblyn, G. V. F. Seaman and H. Walter, J. Colloid Interface Sci., 1984, 102, 1 CrossRef.
- C. A. Haynes, J. Carson, H. W. Blanch and J. M. Prausnitz, AIChE J., 1997, 37, 1401.
Footnote |
| † The HTML version of this article has been enhanced with colour images. |
| This journal is © The Royal Society of Chemistry 2007 |