Electrosynthesis of phenyl-2-propanone derivatives from benzyl bromides and acetic anhydride in an unsupported micro-flow cell electrolysis process†
Ping
He
a,
Paul
Watts
a,
Frank
Marken
b and
Stephen J.
Haswell
*a
aDepartment of Chemistry, University of Hull, Hull, UK HU6 7RX. E-mail: s.j.haswell@hull.ac.uk
bDepartment of Chemistry, University of Bath, Bath, UK BA2 7AY
First published on 3rd November 2006
Abstract
A simple process for the synthesis of phenyl-2-propanone is described based on a one-step electrochemical acylation reaction, involving the direct electroreductive coupling of benzyl bromides and acetic anhydride in a micro-flow electrolysis cell, equipped with micro-gap Pt electrodes. The technique offered yields typically in excess of 80% with corresponding high levels of product selectivity. The electrochemical process was also scaled-up by connecting four identical micro electrochemical cells in parallel to increase product throughput.
Phenyl-2-propanone, commonly referred to as P2P, is probably the most popular intermediate for the manufacture of amphetamine and methamphetamine,1 and represents a versatile intermediate for the synthesis of pharmaceuticals, agrochemicals and fragrances. Due to the relatively simple structure of the compound and because of its common use,1 a number of synthetic routes for its production have been developed. Most of these methods require the presence of a catalyst based on organometallic complexes,2 metal acetates,3 metal halides,4 or Grignard reagents5 to give overall yields of up to 70%. There remains however scope for greener and cleaner methods based, for example, on electrochemical technology to be more effectively exploited. In 1977, Shono6 described a novel electrosynthesis process based on the reduction of benzyl chlorides in the presence of carboxylic acid chlorides in acetonitrile or N,N-dimethylformamide media, and using a conventional two compartment cell with a ceramic diaphragm and 1 M supporting electrolyte. Yields varied between 29 and 73% depending on the starting materials. In 1986, a patent7a reported a process for the synthesis of P2P by electrochemical reduction of benzyl chlorides in the presence of acetic anhydride using an undivided electrolysis cell equipped with a sacrificial anode (i.e. Mg, Al, Zn), organic solvent (N,N-dimethylformamide, acetonitrile, tetrahydrofuran), and supporting electrolyte to give yields of 55% to 64%. In 1994, this process was further modified to use the electrochemical arylation of α-chloroketones with arylhalides in the presence of a catalytic nickel complex.7c All of these cited electrochemically based processes suffer, however, from complicated work up and generate only modest yields.
Micro reactor methodology has been shown to have numerous practical advantages (when compared with batch reactors),8 including a safe operating environment, good process control, and the capability to scale-up for industrial production. In addition electrosyntheses in micro reactors has been shown to offer higher yields, in the absence of a supporting electrolyte,9 which reduces costly work up and purification steps.
In this present study we describe a simple and clean process for the synthesis of P2P based on a one-step electrochemical acylation reaction by direct electroreductive coupling of benzyl bromides and acetic anhydride in a micro-flow electrolysis cell equipped with micro-gap Pt electrodes. The reaction occurs in DMF solvent without supporting electrolyte to generate excellent yields of the products when compared with conventional synthetic methods. Notable benefits of this novel electrochemical process include (i) simple operation, (ii) no need for electrolytes, (iii) minimum product work-up, and (iv) high yield and selectivity of products.
Initially, the acylation reaction of benzyl bromide with acetic anhydride was studied by cyclic voltammetry at conventional Pt-disc (diameter 0.5 mm) and micro Pt-disc electrodes (diameter 25 µm) to establish the reaction mechanism. The electroreduction of benzyl bromide is chemically irreversible, leading to the formation of either toluene via a two-electron reduction10 or dibenzyl formally via a one-electron reduction. Fig. 1 shows cyclic voltammograms obtained in DMF for (i) the reduction of acetic anhydride, (ii) the reduction of benzyl bromide, and (iii) the reduction of benzyl bromide in the presence of excess acetic anhydride.
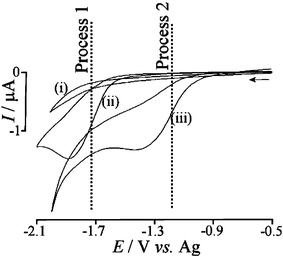 | ||
| Fig. 1 Cyclic voltammograms (scan rate of 0.1 V s−1) obtained at a 0.5 mm diameter platinum disc electrode immersed in 0.1 M n-Bu4NBF4–DMF for (i) 60 mM acetic anhydride, (ii) 3 mM benzyl bromide, and (iii) 3 mM benzyl bromide in the presence of 60 mM acetic anhydride. | ||
The irreversible reduction of benzyl bromide (Process 1) occurs as a two-electron process (see ESI†). In the presence of acetic anhydride, a new reductive peak appears (Process 2) at more positive potential position. The peak current for Process 2 increases with increasing amounts of acetic anhydride (for 15 mM to 60 mM) whilst the peak current for Process 1 gradually decreases. The overall mechanism remains a two-electron transfer, with Process 2 being observed only at platinum electrode surfaces and not at glassy carbon or gold (see ESI†). The ratio of peak currents for Processes 1 and 2 is scan rate dependent, consistent with a fast preceding chemical step coupled to electron transfer at the platinum surface. Acetyl from acetic anhydride is likely to act as a “trap” for a benzyl anion intermediate formed at the platinum electrode surface.
Preparative micro reactor electrolysis was conducted in a rectangular cavity micro-flow cell (see Fig. 2) with products being determined off-line by using GC/MS and 1H-NMR. The reaction medium, containing 5 mM benzyl bromide in DMF with varied amount of acetic anhydride, was continuously pumped through the cell, in which two platinum electrodes with a working area of 45 mm2 each were positioned with an inter-electrode gap of 160 µm to produce a 7.2 µl cell volume.
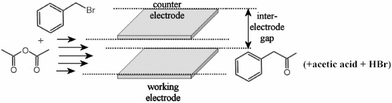 | ||
| Fig. 2 Schematic representation of the acylation reaction during micro reactor electrosynthesis. A flow of reagents through a rectangular cavity with working and counter electrode facing each other results in the formation of products. | ||
The electro-acylation reactions were conducted galvano-statically and product samples were collected for a 30 minute period. Table 1 summarizes the conversion and product distribution for the range of conditions employed in this study.
| Entry | Current/mA |
|
AA/BBb (mol/mol) | Conv. (%)c | Distribution (%) | ||
|---|---|---|---|---|---|---|---|
| R1 | R2 | P2Pd | DBre | ||||
| a 5 mM benzyl bromides, acetic anhydride concentration as shown in the Table, electrode gap is 160 µm, electrode area 45 mm2, flow rate 10 µl min−1 corresponding to 43 s contact time. b Molar concentration ratio. c The conversion was determined based on the quality of benzyl bromide before and after reaction using n-decane as an internal standard. d P2P represents P2P or its derivatives. e DBr is debromination yield for benzyl bromides. f Side products include debromination of benzyl bromide (16%) and dimer formation (10%). | |||||||
| 1 | 0.8 | H | H | 10 | 87 | 61 | 26f |
| 2 | 0.8 | H | H | 20 | 85 | 62 | 23 |
| 3 | 1.1 | H | H | 20 | 92 | 66 | 26 |
| 4 | 1.1 | H | H | 40 | 90 | 81 | 9 |
| 5 | 1.1 | CH3 | H | 40 | 93 | 87 | 6 |
| 6 | 1.1 | H | CH3 | 40 | 98 | 96 | 2 |
| 7 | 1.3 | CH3O | H | 40 | 99 | 83 | 16 |
| 8 | 1.1 | Br | H | 40 | 73 | 51 | 22 |
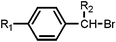
From Table 1 it can be seen (Entry 1–4) that the conversion and product distribution are dependent on the molar ratio of benzyl bromide to acetic anhydride and the applied current (or potential). Voltages between 5–5.4 V were required to obtain sufficiently high levels of conversion (>85%) in most cases. The best result obtained was 81% of phenyl-2-propanone with 9% of toluene at a flow rate of 10 µl min−1 (corresponding to 43 s contact time, see Entry 4). Lower ratios of acetic anhydride to benzyl bromide led to the formation of more toluene (Entry 2–3), and even the formation of the dibenzyl product (Entry 1).
Other benzyl bromide derivatives such as 1-phenylethyl bromide, 4-methylbenzyl bromide, 4-methoxybenzyl bromide, and 4-bromobenzyl bromide were also examined (see Entry 5–8 in Table 1) for the acylation reaction with acetic anhydride. It is noted that the presence of Br– and CH3O– groups on the benzyl bromides promote the formation of the debromination products (see Entry 7–8), compared to CH3– and H– groups. In contrast, in the presence of an electron donating group (see Entry 5–6) yields are improved. The formation of bromine due to oxidation of bromide (as a follow up anodic process) was not observed, presumably due to the limited overlap of diffusion layers within the flow cell. The diffusion layer thickness for the process can be estimated based on eqn (1).
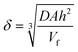 | (1) |
In this equation the diffusion layer thickness is obtained based on the diffusion coefficient D, the electrode area A, the half height of the cell h, and the volume flow rate Vf. For a diffusion coefficient of 10−9 m2 s−1 and under conditions employed here, the diffusion layer thickness is estimated as δ = 120 µm, which is approaching the inter-electrode distance.
Current efficiencies for all processes are typically around 20–25%, consistent with an overall transfer of 4 electrons per benzyl bromide, but background currents in the presence of acetic anhydride are likely to be responsible for the low yield (see Fig. 1). This observation is consistent with literature reports.7a,b The electrochemical process was also scaled-up by connecting four identical micro electrochemical cells in parallel. In this case, a similar level of product yield was obtained with a four-fold increase in the quality of the product formation. In summary, micro-flow electrosynthesis offers a surprisingly simple and low waste access to phenyl-2-propanone derivatives which is readily optimized and can be scaled-up. It is very likely that in future a wider range of chemical processes will be identified to be suitable for this kind of simple and clean micro-reactor technology.
Acknowledgements
We thank EPSRC for funding (Project No. GR/S34106)Notes and references
- A. C. Allen and T. S. Cantrell, Synthetic reductions in clandestine amphetamine and methamphetamine laboratories: A Review, Forensic Science International, Elsevier, 1989, vol. 42, p. 183 Search PubMed.
- (a) R. Ballini, Synthesis, 1994, 723 CrossRef CAS; (b) S. Inaba and R. D. Rieke, Tetrahedron Lett., 1983, 24, 2451 CrossRef CAS; (c) S. Inaba and R. D. Rieke, J. Org. Chem., 1985, 50, 1373 CrossRef CAS.
- (a) M. E. Kurz, V. Baru and P. N. Nguyen, J. Org. Chem., 1984, 49, 1603 CrossRef CAS; (b) A. C. Allen, M. L. Stevensen, S. M. Nakamura and R. A. Ely, J. Forensic Sci., 1992, 37, 301 CAS.
- (a) K. Lee and D. Y. Oh, Tetrahedron Lett., 1988, 29, 2977 CrossRef CAS; (b) K. Okabe, T. Ohwada, T. Ohta and K. Shudo, J. Org. Chem., 1989, 54, 733 CrossRef CAS.
- (a) P. Canonne, G. B. Foscolos and G. Lemay, Tetrahedron Lett., 1980, 21, 155 CrossRef CAS; (b) K. Okabe, T. Ohwada, T. Ohta and K. Shudo, J. Org. Chem., 1972, 37, 3369 CrossRef.
- T. Shono, Chem. Lett., 1977, 1021 CrossRef CAS.
-
(a)
M. D. Moingeon and J. Chaussard, US Pat., 4
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 629541, 1986 Search PubMed;
(b) E. D'Incan, S. Silbille, J. Périchon, M. D. Moingeon and J. Chaussard, Tetrahedron Lett., 1986, 27, 4175 CrossRef CAS;
(c) M. Durandetti, S. Sibille, J.-Y. Nédélec and J. Périchon, Synth. Commun., 1994, 24, 145 CrossRef CAS.
629541, 1986 Search PubMed;
(b) E. D'Incan, S. Silbille, J. Périchon, M. D. Moingeon and J. Chaussard, Tetrahedron Lett., 1986, 27, 4175 CrossRef CAS;
(c) M. Durandetti, S. Sibille, J.-Y. Nédélec and J. Périchon, Synth. Commun., 1994, 24, 145 CrossRef CAS. - (a) S. Taghavi-Moghadam, A. Kleemann and K. G. Golbig, Org. Process Res. Dev., 2001, 5, 652 Search PubMed; (b) T. Kawaguchi, H. Miyata, K. Ataka, K. Mae and J. Yoshida, Angew. Chem., Int. Ed., 2005, 44, 2413 CrossRef CAS.
- (a) C. A. Paddon, G. J. Pritchard, T. Thiemann and F. Marken, Electrochem. Commun., 2002, 4, 825 CrossRef CAS; (b) R. Horcajada, M. Okajima, S. Suga and J. Yoshida, Chem. Commun., 2005, 1303 RSC; (c) P. He, P. Watts, F. Marken and S. J. Haswell, Electrochem. Commun., 2005, 7, 918 CAS; (d) P. He, P. Watts, F. Marken and S. J. Haswell, Angew. Chem., Int. Ed., 2006, 45, 4146 CrossRef CAS.
- (a) J. Grimshaw, Electrochemical Reactions and Mechanisms in Organic Chemistry, Elsevier, Amsterdam, 2000, pp. 98–103 Search PubMed; (b) Organic Electrochemistry, ed. H. Lund and O. Hammerich, Marcel Dekker, New York, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, P2P syntheses described in literature and characterization. See DOI: 10.1039/b610415k |
| This journal is © The Royal Society of Chemistry 2007 |