Can ionic liquids dissolve wood? Processing and analysis of lignocellulosic materials with 1-n-butyl-3-methylimidazolium chloride
Diego A.
Fort
a,
Richard C.
Remsing
b,
Richard P.
Swatloski
cd,
Patrick
Moyna
*a,
Guillermo
Moyna
*b and
Robin D.
Rogers
*c
aFacultad de Química, Universidad de la República, Avenida General Flores 2124, Montevideo 11800, Uruguay. E-mail: pmoyna@fq.edu.uy
bDepartment of Chemistry & Biochemistry, University of the Sciences in Philadelphia, 600 South 43rd Street, Philadelphia, PA 19104, USA. E-mail: g.moyna@usip.edu
cCenter for Green Manufacturing and Department of Chemistry, The University of Alabama, Tuscaloosa, AL 35487, USA. E-mail: rdrogers@bama.ua.edu
d525 Solutions, Inc., P.O. Box 861395, Tuscaloosa, AL 35486, USA
First published on 17th October 2006
Abstract
The bulk of the cellulose currently employed by industry is isolated from wood through Kraft pulping, a process which traditionally involves a barrage of environmentally detrimental chemicals and is undeniably ‘non-green.’ In this report we present a simple and novel alternative approach for the processing of lignocellulosic materials that relies on their solubility in solvent systems based on the ionic liquid (IL) 1-n-butyl-3-methylimidazolium chloride ([C4mim]Cl). Dissolution profiles for woods of different hardness are presented, making emphasis on the direct analysis of the cellulosic material and lignin content in the resulting liquors by means of conventional 13C NMR techniques. We also show that cellulose can be readily reconstituted from the IL-based wood liquors in fair yields by the addition of a variety of precipitating solvents. Spectroscopic and thermogravimetric studies indicate that the polysaccharide obtained in this manner is virtually free of lignin and hemicellulose and has characteristics that are comparable to those of pure cellulose samples subjected to similar processing conditions.
Introduction
Wood is one of the most important biorenewable resources available to Man, with well-known uses as fuel, building, and manufacturing material.1,2 In addition, it is the primary source of cellulose for use in the paper, fiber, paint, membrane, and polymer industries. Among the different approaches currently employed in the production of cellulose from wood, Kraft pulping methods are by far the most heavily used.3,4 Briefly, the process involves the semi-selective chemical degradation of the lignin /hemicellulose matrix in wood by treatment with solutions of sodium hydroxide and sodium sulfide at high temperatures and pressures. This selectivity results in high yields of cellulose fibers that are stronger and require less bleaching than those obtained by other comparable methods. Despite its popularity and efficiency, Kraft pulping involves the use of a variety of toxic and hazardous chemicals and generates large amounts of air and water pollutants.3,4 In view of the fact that the annual production of wood pulp nears 200 million tons worldwide,2 roughly half of which comes from North America,2,4 the environmental impact of the paper, pulp, and associated industries has become a major global concern. A number of improvements to traditional pulping methods, together with elemental chlorine free (ECF) and totally chlorine free (TCF) bleaching protocols introduced in the 1990s,3,4 have considerably reduced pollution levels generated by modern Kraft pulp mills. In addition, cleaner and more efficient pulp delignification strategies based on aqueous biphasic extraction systems, such as the clean fractionation process and methods relying on organosolv or polyethylene glycol (PEG),5–7 have been demonstrated in recent years. Notwithstanding these advances, the constant tightening of policies affecting this sector and industry self-regulation initiatives have made the search for ‘greener’ wood processing technologies a never ending endeavor.The enormous promise of ionic liquids (ILs) as environmentally-friendly materials is now well known, and their numerous applications have been extensively documented.8,9 ILs have tunable physicochemical properties, usually negligible vapor pressures and, in general, thermal stability over a wide range of temperatures. As a result, they have been used successfully to replace traditional solvents employed in a variety of synthetic and manufacturing processes, and have the potential to reduce the current dependency of industry on volatile organic compounds (VOCs).8,9 In addition to their role as alternative reaction and extraction media, we and others have shown that certain ILs can dissolve polysaccharides and other biopolymers very efficiently.10–15 Perhaps the most salient results in this regard have been those obtained with 1-n-butyl-3-methylimidazolium chloride ([C4mim]Cl, Fig. 1a). This IL dissolves cellulose with no prior derivatization in concentrations of up to 300 g L–1, thus facilitating the development of ‘greener’ technologies for the production of cellulose-based materials.10,16 As we have recently reported, the solvation of carbohydrates in [C4mim]Cl involves the formation of hydrogen bonds between the non-hydrated chloride ions of the IL and the sugar hydroxyl protons in a 1:1 stoichiometry.17 In the case of cellulose, these interactions disrupt the extensive hydrogen bonding network of the polymer and lead to its dissolution. Additionally, we have shown that following minimal sample preparation [C4mim]Cl is capable of fully dissolving structured polysaccharide-based natural matrices, such as those found in fruits and grains, allowing for their composing carbohydrates to be readily extracted and analyzed.18 Taking its unique properties as a cellulose solvent also into account, these latter findings prompted us to explore the use of this IL in the development of much needed alternative methods for the extraction and processing of polysaccharides from wood and other forms of lignocellulosic biomass .19
![Structures of [C4mim]Cl (a), cellulose (b), and lignin (c).](/image/article/2007/GC/b607614a/b607614a-f1.gif) | ||
| Fig. 1 Structures of [C4mim]Cl (a), cellulose (b), and lignin (c). | ||
As discussed herein, our results indicate that solvent systems based on [C4mim]Cl are indeed capable of partially dissolving wood. In addition, we demonstrate that celluloses which are virtually free of lignin and hemicellulose can be easily reconstituted from the resulting IL-based wood extracts, and show that the physicochemical properties of the recovered materials are comparable to those of pure cellulose samples subjected to similar processing conditions.
Results and discussion
The dissolution in [C4mim]Cl of woods of varying hardness, including pine, poplar, eucalyptus, and oak, was studied using an approach based on previously reported methods.18,20 For each sample, 5 wt% suspensions of dried and finely divided wood chips in a solution of the IL and 15 wt% DMSO-d6 were stirred at 100 °C for periods ranging from 2 to 24 h. As we have shown, the use of small amounts of DMSO-d6 as a co-solvent has no noticeable effect on the solubility of carbohydrates in [C4mim]Cl, reduces the viscosity of the mixtures, and, more importantly, allows for the resulting solubilized materials to be analyzed directly with conventional 13C NMR techniques.18,20 In all cases, swelling of the wood particles was observed immediately after they came in contact with the IL-based solvent. As time progressed, the coloration and viscosity of the solutions increased and the suspended particles became smaller, indicating that dissolution was taking place. However, none of the samples dissolved completely even after extended periods of time. This is not surprising in view of the fact that the structural matrix of the cell walls of higher plants, made up primarily of tightly associated cellulose and lignin chains (Fig. 1),1 is significantly more complex than that of the amyloplasts from fruits and grains that were considered in our earlier studies. Following the removal of undissolved particulate matter, the compositions of the resulting wood extracts, or wood liquors, were inspected using 13C NMR spectroscopy.18,20The 13C spectrum of the liquor obtained after dissolving pine wood for 6 h in [C4mim]Cl/DMSO-d6, presented in Fig. 2a, serves to illustrate the most salient aspects of our analyses. Comparison with the spectrum of microcrystalline cellulose (MCC) recorded under identical conditions reveals clearly that this polysaccharide is present in the IL-based wood liquor (Fig. 2b). Of note are the signals at 103.1, 80.1, 76.2, 75.9, 74.7, and 61.0 ppm, all of which are well-resolved and correspond, respectively, to the resonances of the C-1, C-4, C-5, C-3, C-2, and C-6 carbons of cellulose.20 Similarly, the signal at 57.2 ppm in the spectrum of the liquor correlates with the aromatic OMe 13C resonance observed in [C4mim]Cl/DMSO-d6 solution for Indulin AT (Fig. 2c), a desalted lignin preparation that closely represents the naturally-occurring phenylpropanoid polymer. Though less significant, signals at 103.6, 97.7, 93.0, 80.7, 77.8, 71.4, and 61.9 ppm can be attributed to low molecular weight cellulose oligomers (i.e., γ-celluloses),20 and indicate that minor degradation of the polysaccharide occurs under these conditions. Finally, considerably weaker signals are also observed at ∼170 and ∼22 ppm (data not shown). Albeit tentatively, these can be assigned to the carbonyl and methyl carbons of acetate groups present in hemicelluloses by comparison with 13C CP/MAS NMR data reported for these heteropolysaccharides.21 Overall, the data presented above prove conclusively that there is partial dissolution of the wood samples in the IL-based solvent system.
![13C NMR spectra of the extract obtained after dissolving pine wood in [C4mim]Cl/DMSO-d6 for 6 h at 100 °C (a), and of 5 wt% solutions of MCC (b) and Indulin AT (c) in the same solvent system.](/image/article/2007/GC/b607614a/b607614a-f2.gif) | ||
| Fig. 2 13C NMR spectra of the extract obtained after dissolving pine wood in [C4mim]Cl/DMSO-d6 for 6 h at 100 °C (a), and of 5 wt% solutions of MCC (b) and Indulin AT (c) in the same solvent system. | ||
The next step in our study involved the estimation of the cellulosic material and lignin extraction levels as a function of dissolution time, or dissolution profiles, for the various types of wood examined. As was stated earlier, the IL-based wood liquors become more viscous and their coloration intensifies with time, a qualitative observation which can be rationalized by an increase in the concentration of polymeric materials in solution.10 This assumption can be confirmed by cursory inspection of the 13C NMR spectra of [C4mim]Cl/DMSO-d6 liquors obtained at different dissolution times, such as those shown in Fig. 3 for poplar wood extracts. Since these spectra were recorded under analogous experimental conditions, the progressive improvement in the signal-to-noise ratio of the cellulose and lignin resonances is a direct reflection of the increase in the concentration of these polymers in the liquor as a function of dissolution time. Further analysis of the data shown in Fig. 3 also reveals that the levels of γ-celluloses grow with time, and although they are still minor components of the solution after 24 h their concentration becomes non-negligible. The remaining types of wood considered in the study followed similar dissolution trends to the one discussed here for poplar.
![13C NMR spectra of the extracts obtained after dissolving poplar wood in [C4mim]Cl/DMSO-d6 for periods of 2 (a), 6 (b), 12 (c), and 24 (d) h at 100 °C. Vertical scalings were adjusted to make signal intensities comparable among all spectra.](/image/article/2007/GC/b607614a/b607614a-f3.gif) | ||
| Fig. 3 13C NMR spectra of the extracts obtained after dissolving poplar wood in [C4mim]Cl/DMSO-d6 for periods of 2 (a), 6 (b), 12 (c), and 24 (d) h at 100 °C. Vertical scalings were adjusted to make signal intensities comparable among all spectra. | ||
In order to quantitate cellulosic material and lignin extraction levels, the average of the integrals of the cellulose and cellulose oligomers C-1 and C-6 signals in the 105–92 and 63–59 ppm range, respectively, and the integral of the lignin aromatic OMe 13C resonance at ∼57 ppm, were computed for each wood liquor spectrum. These values were then divided by the integrals of the corresponding signals observed in 5 wt% solutions of MCC and Indulin AT in [C4mim]/DMSO-d6, using the areas of the IL 13C signals in each spectrum as internal normalization factors. Since the initial concentration of wood used in all dissolution experiments was the same as that of the cellulose and lignin reference solutions, these ratios represent the cellulosic material and lignin wt% extraction levels achieved for each sample. This approach was therefore employed to generate dissolution profiles in the IL-based solvent system as a function of time for the four types of wood investigated (Fig. 4). It is important to point out that the poor to modest signal-to-noise ratio observed in the 13C NMR spectra of the extracts can lead to sizable integration errors, and as a result the extraction levels estimated in this manner have uncertainties of up to ±5 wt%.
![Cellulosic material (solid bars) and lignin (dashed bars) extraction profiles in [C4mim]Cl/DMSO-d6 at 100 °C for the different wood samples considered as a function of time.](/image/article/2007/GC/b607614a/b607614a-f4.gif) | ||
| Fig. 4 Cellulosic material (solid bars) and lignin (dashed bars) extraction profiles in [C4mim]Cl/DMSO-d6 at 100 °C for the different wood samples considered as a function of time. | ||
Analysis of the profiles shown in Fig. 4 yields valuable information regarding the wood dissolution process. First, and in agreement with the differences in morphological properties and complexity of their lignocellulosic matrices, extraction levels for hardwoods are lower than for softwoods. For example, 44 wt% of the cellulosic materials present in pine wood can be extracted by the IL-based solvent in 12 h, but extraction levels reach only 35 wt% for oak in the same time period. In addition, the cellulosic material to lignin wt% ratio across the dissolution profiles is largely constant and roughly 2:1. This figure correlates well with the reported chemical composition of wood,1 and suggests that the [C4mim]Cl/DMSO-d6 mixture extracts the two polymers from the wood fibers at approximately equal rates. Finally, the cellulosic material extraction levels after 24 h are, on average, 45 wt%. If this value is again compared with the wt% chemical make-up of wood,1 our results indicate that the IL-based solvent system is capable of extracting the majority of the cellulose present in the samples. Indeed, we found that continuing the dissolution process for longer periods does not lead to an increase in the concentration of cellulosic materials or lignin in the liquors. On the other hand, and as evidenced by a noticeable increase in the intensities of resonances corresponding to γ-celluloses, contact times beyond 24 h lead to substantial polymer degradation.
Cellulose recovery from the IL-based wood liquors was then investigated. We have previously shown that the polysaccharide can be reconstituted from [C4mim]Cl solutions by the addition of precipitating solvents and solvent mixtures miscible with the IL,10,16 and the suitability of methods based on these techniques was explored. Analysis of the dissolution profiles for softwoods presented above indicates that good levels of cellulosic material extraction can be achieved with minimal degradation between 12 and 24 h. Thus, solutions for cellulose reconstitution studies were prepared by dissolving, or pulping, pine wood in the [C4mim]Cl/DMSO mixture for 16 h at 100 °C. In order to resemble more closely non-ideal conditions encountered in an industrial setting, the pulping experiments were carried out on a multi-gram scale and using coarse wood shavings with no preconditioning. Barring an initial evolution of steam from the heterogeneous slurries, the dissolution of the untreated wood fibers proceeded as described earlier, and clear amber colored liquors were obtained after removal of insoluble particles. The reconstitution of the cellulosic materials was achieved through the addition and vigorous mixing of one to two volumes of the precipitating solvents, which included 1:1 acetone–water solutions, dichloromethane or acetonitrile. In all cases, off-white powdery flocs formed shortly after the coagulant mixed with the wood liquors. The solids were recovered by filtration and washed repeatedly with water and acetone to completely remove [C4mim]Cl and DMSO. Based on the initial concentration of the wood suspensions and the average cellulose content in wood,1 the yields of the reconstitution experiments ranged from 30 to 60 wt%.
As expected, the recovered materials were insoluble in traditional laboratory solvents. On the other hand, they dissolved readily in neat [C4mim]Cl or [C4mim]Cl/DMSO-d6, and 5 wt% solutions suitable for NMR spectroscopy could be prepared in all cases. Comparison of the 13C spectra of the reconstituted samples with that of MCC recorded under identical conditions reveals that they are celluloses of fair purity (Fig. 5). Although γ-cellulose resonances are discernible in all cases, their intensities are considerably lower than in the spectra of the original wood liquors (i.e., Fig. 3d). Furthermore, similar signals are present in the spectrum of MCC, suggesting that these low molecular weight oligomers are the result of minor decomposition of the polysaccharide occurring during sample preparation. Perhaps the most salient feature of the spectra is their lack of signals at ∼57 ppm or ∼170 and ∼22 ppm that could be attributed, respectively, to lignin aromatic OMe or hemicellulose acetate groups. This indicates that these polymers are not present in significant concentrations in the reconstituted cellulose samples. Since lignins and hemicelluloses were observed in the IL-based wood liquors prior to the cellulose precipitation step, both must remain in the coagulation filtrates. While no effort was made to recover these polymers, their presence in the supernatants was confirmed by 13C NMR (data not shown). Similar conclusions regarding the purity of the recovered materials can be reached by comparison of their IR spectra, all of which lack absorption bands that characterize lignin and hemicellulose,22 with that of pure MCC (Fig. 6).
![13C NMR spectra of 5 wt% solutions in [C4mim]Cl/DMSO-d6 of celluloses reconstituted from pine wood liquors using acetone–water (a), dichloromethane (b), and acetonitrile (c), and of MCC (d).](/image/article/2007/GC/b607614a/b607614a-f5.gif) | ||
| Fig. 5 13C NMR spectra of 5 wt% solutions in [C4mim]Cl/DMSO-d6 of celluloses reconstituted from pine wood liquors using acetone–water (a), dichloromethane (b), and acetonitrile (c), and of MCC (d). | ||
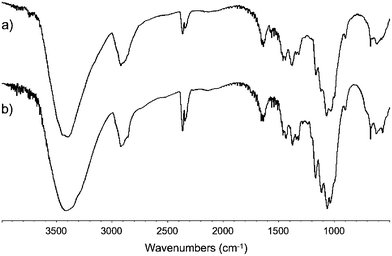 | ||
| Fig. 6 IR spectra of cellulose reconstituted from IL based pine wood liquors by the addition of acetonitrile (a) and of MCC (b). | ||
The thermal decomposition profiles of the recovered celluloses were then determined using thermogravimetric analysis (TGA). Neglecting small initial drops in weight occurring near 100 °C due to evaporation of retained moisture, a single pronounced decomposition event followed by slow loss of mass was observed in all cases. The decomposition onset and maximum decomposition rate temperatures computed from the TGA traces of the different samples, Tdec and Tmax, were in the 257–301 °C and 276–320 °C range, respectively, and their residual char yields above 500 °C were between 12.8 and 21.6 wt%. This range of thermal decomposition parameters is comparable to that determined for MCC samples before and after dissolution in [C4mim]Cl/DMSO and reconstitution with acetone–water (Fig. 7), which had Tdec and Tmax values of 307 and 324 °C and 257 and 269 °C, respectively. While qualitatively, this indicates that the variations in the morphology and degree of polymerization (DP) of the celluloses recovered from the IL-based wood liquors are within those of pure and [C4mim]Cl-processed MCC.23
![Thermal decomposition profiles of cellulose reconstituted from IL based pine wood liquors by the addition of dichloromethane (a), and of samples of MCC before (b) and after (c) dissolution in [C4mim]Cl/DMSO and regeneration with acetone–water.](/image/article/2007/GC/b607614a/b607614a-f7.gif) | ||
| Fig. 7 Thermal decomposition profiles of cellulose reconstituted from IL based pine wood liquors by the addition of dichloromethane (a), and of samples of MCC before (b) and after (c) dissolution in [C4mim]Cl/DMSO and regeneration with acetone–water. | ||
As a final assessment of their physical properties, we explored the use of the regenerated samples in the preparation of cellulose-based membranes following methods described earlier by us.24 Briefly, 5 wt% solutions of the reconstituted celluloses redissolved in [C4mim]Cl were prepared, cast onto glass plates as thin films, and perfused extensively with water. In all cases, highly flexible membranes identical in all respects to those prepared analogously using pure MCC were obtained. In contrast, all attempts to cast membranes directly from the IL-based wood extracts proved unsuccessful, suggesting that the presence of lignins and/or hemicelluloses inhibits the formation of structured cellulose hydrogels.
Conclusions
The results discussed above demonstrate that solvent systems based on [C4mim]Cl are capable of partially dissolving untreated wood, and show that celluloses with purities, physical properties, and processing characteristics comparable to those of pure cellulose samples subjected to similar treatment can be easily recovered from the resulting solutions. In contrast to wood delignification methods that rely on the insolubility of cellulose, the IL has the ability to dissolve lignin and polysaccharides simultaneously. Thus, the intricate network of non-covalent interactions between these polymers is effectively disrupted after dissolution, leading to celluloses which are devoid of lignin and hemicellulose upon regeneration. Furthermore, the fact that the wood components are in [C4mim]Cl solution should, in principle, allow for their in situ derivatization. For example, cellulose could be acylated or carbanilated directly in the IL-based wood liquors following recently reported procedures,11,25 drastically reducing the number of steps currently required to produce these derivatives from raw materials.While our findings are encouraging, several aspects of the novel methodology described here need to be addressed before it can be considered as an alternative ‘green’ strategy for the extraction of cellulose from lignocellulosic materials. First, the effects of dissolution time and temperature on the crystallinity and DP of the celluloses obtained through this approach were not studied, and these should be rigorously quantitated. In addition, [C4mim]Cl has been shown to display moderate toxicity,26 and use of this IL on large scales must be combined with efficient recycling protocols.27 The suitability as wood solvents of [C2mim]Cl, an IL with a slightly higher melting point but significantly less toxicity than [C4mim]Cl, and other imidazolium chlorides known to solubilize cellulose, could be evaluated as well.11,25 Similarly, the use of DMSO as a dissolution co-solvent should be minimized or avoided. In our case, DMSO-d6 and DMSO were employed solely to reduce the viscosity of the IL-based wood liquors and facilitate their handling and analysis. Preliminary results indicate that similar reductions in viscosity can be achieved with PEG, a solvent which we have successfully used in the development of cleaner methods for the delignification of wood pulps.7 Finally, the energy requirements of the process need to be determined and compared with those of established cellulose extraction and purification protocols. Work on these areas is well under way, and the results from our ongoing investigations will be reported in due course.
Experimental
Materials
Pine (Pinus spp.), poplar (Populus spp.), eucalyptus (Eucalyptus spp.), and oak (Quercus spp.) wood shavings were procured from local woodshops. With the exception of the materials employed in pulping/reconstitution experiments, the samples were finely divided with a mortar and pestle and dried to constant weight under vacuum before use. The [C4mim]Cl used in all the studies was prepared following reported procedures.28 Samples of Indulin AT were obtained from Westvaco (Charleston, SC, USA). DMSO-d6 (99.9% D) was purchased from Cambridge Isotope Laboratories (Cambridge, MA, USA). All other solvents and reagents were obtained from Sigma–Aldrich (St. Louis, MO, USA).Wood dissolution profiles
5 wt% suspensions of wood chips in IL–DMSO-d6 were prepared by combining 100 mg of the different samples, 1.6 g of [C4mim]Cl, and 300 mg of DMSO-d6 in test tubes followed by thorough vortexing. The heterogeneous mixtures were then heated at 100 °C for periods of either 2, 6, 12, or 24 h with constant stirring, and subsequently filtered through glass wool to remove undissolved material. The resulting clear liquors were transferred to 5 mm NMR tubes and used in the determination of wood dissolution profiles, as detailed above.Wood pulping and cellulose reconstitution experiments
1 g of untreated pine wood shavings (∼5 mm × 5 mm in size, 5–10 wt% moisture content) were suspended in 19 g of a [C4mim]Cl/DMSO solution (84:16 wt%), and pulped at 100 °C for 16 h with the aid of a magnetic stirrer. The mixture was then filtered through a heated Büchner funnel fitted with a 20 µm Magna® nylon membrane (Osmonics/MSI, Westborough, MA, USA) to remove undissolved solids. Cellulose samples were reconstituted from the resulting clear, amber-colored, viscous liquor as flocs by rapid mixing with ∼50 mL of either 1:1 acetone–water, dichloromethane, or acetonitrile. The powdery off-white solids were then filtered, washed repeatedly with acetone and water, and dried overnight in a vacuum oven at 100 °C prior to their use in 13C NMR, IR, and TGA experiments.NMR and IR spectroscopy
Samples used in the determination of wood dissolution profiles were obtained as described in the previous section. The 5 wt% solutions of MCC, Indulin AT, and reconstituted cellulose samples in [C4mim]/DMSO-d6 used in NMR experiments were prepared as reported earlier.18,20 Proton-decoupled 13C spectra were collected at 90 °C on a Bruker AVANCE 400 NMR spectrometer equipped with a 5 mm BBO probe operating at a 13C frequency of 100.61 MHz. A total of 20![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 scans were collected in all cases, and spectra were processed with a 10 Hz line-broadening factor.
000 scans were collected in all cases, and spectra were processed with a 10 Hz line-broadening factor.
IR spectra of the cellulose samples were recorded on a Thermo-Electron Nicolet Avatar 370 DTGS FT-IR spectrophotometer using KBr pellets.
Thermogravimetric analysis
The thermal decomposition profiles of the reconstituted cellulosic materials were determined using a TA Instruments 2950 thermogravimetric analyzer (New Castle, DE, USA). Each sample was analyzed in a platinum pan with nitrogen as the purge gas. In all experiments the temperature was increased from 30 to 700 °C at a constant rate of 10 °C min–1.Acknowledgements
Funding from the NSF CCLI-A&I program (Grant DUE-9952264), the Camille and Henry Dreyfus Foundation, and DHC Analysis is acknowledged (GM, RCR, DAF, and PM). RDR and RPS recognize support from BASF Corporation.References
- Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, New York, 4th edn., 1998, vol. 25, pp. 627–664 Search PubMed.
- FAO Yearbook of Forest Products 2003, FAO Forestry Series No. 38 and Statistics Series No. 185, Food and Agriculture Organization of the United Nations, Rome, Italy, 2005 Search PubMed.
- Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, New York, 4th edn., 1996, vol. 20, pp. 494–582 Search PubMed.
- Profile of the Pulp and Paper Industry, EPA Office of Compliance Sector Notebook EPA/310-R-02-002, US Environmental Protection Agency, Washington, DC, 2nd edn., 2002 Search PubMed.
-
S. K. Black, B. R. Hames and M. D. Myers, US Pat., 5730837, 1998 Search PubMed.
- T. J. McDonough, Tappi J., 1993, 76, 186–193 Search PubMed.
- H. D. Willauer, J. G. Huddleston, M. Li and R. D. Rogers, J. Chromatogr., B: Biomed. Appl., 2000, 743, 127–135 CrossRef CAS.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef.
- Ionic Liquids IIIA/B: Fundamentals, Progress, Challenges, and Opportunities, ed. R. D. Rogers and K. R. Seddon, ACS Symp. Ser. 901/902, American Chemical Society, Washington, DC, 2005 Search PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS.
- J. Wu, J. Zhang, H. Zhang, J. He, Q. Re and M. Guo, Biomacromolecules, 2004, 5, 266–268 CrossRef CAS.
- Q. Liu, M. H. A. Janssen, F. van Rantwijk and R. A. Sheldon, Green Chem., 2005, 7, 39–42 RSC.
- D. M. Phillips, L. F. Drummy, D. G. Conrady, D. M. Fox, R. R. Naik, M. O. Stone, P. C. Trulove, H. C. De Long and R. A. Mantz, J. Am. Chem. Soc., 2004, 126, 14350–14351 CrossRef CAS.
- H. Xie, S. Li and S. Zhang, Green Chem., 2005, 7, 606–608 RSC.
- H. Xie, S. Zhang and S. Li, Green Chem., 2006, 8, 630–633 RSC.
-
R. P. Swatloski, R. D. Rogers and J. D. Holbrey, US Pat., 6824599, 2004 Search PubMed.
- R. C. Remsing, R. P. Swatloski, R. D. Rogers and G. Moyna, Chem. Commun., 2006, 1271–1273 RSC.
- D. A. Fort, R. P. Swatloski, P. Moyna, R. D. Rogers and G. Moyna, Chem. Commun., 2006, 714–716 RSC.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef.
- J. S. Moulthrop, R. P. Swatloski, G. Moyna and R. D. Rogers, Chem. Commun., 2005, 1557–1559 RSC.
- W. Kolodziejski, J. S. Frye and G. E. Maciel, Anal. Chem., 1982, 54, 1419–1424 CrossRef CAS.
- T. P. Abbott, D. M. Palmer, S. H. Gordon and M. O. Bagby, J. Wood Chem. Technol., 1988, 8, 1–28 CrossRef CAS.
- M. E. Calahorra, M. Cortázar, J. I. Eguiazábal and G. M. Guzmán, J. Appl. Polym. Sci., 1989, 37, 3305–3314 CrossRef CAS.
-
(a)
J. D. Holbrey, S. K. Spear, M. B. Turner, R. P. Swatloski and R. D. Rogers, US Pat., 6808557, 2004 Search PubMed;
(b) M. B. Turner, S. K. Spear, J. D. Holbrey and R. D. Rogers, Biomacromolecules, 2004, 5, 1379–1384 CrossRef CAS;
(c) M. B. Turner, S. K. Spear, J. D. Holbrey, D. T. Daly and R. D. Rogers, Biomacromolecules, 2005, 6, 2497–2502 CrossRef CAS.
- S. Barthel and T. Heinze, Green Chem., 2006, 8, 301–306 RSC.
- R. P. Swatloski, J. D. Holbrey, S. B. Memon, G. A. Caldwell, K. A. Caldwell and R. D. Rogers, Chem. Commun., 2004, 668–669 RSC.
- K. E. Gutowski, G. A. Broker, H. D. Willauer, J. G. Huddleston, R. P. Swatloski, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2003, 125, 6632–6633 CrossRef CAS.
- J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156–164 RSC.
| This journal is © The Royal Society of Chemistry 2007 |