Novel chemical recycling of polycarbonate (PC) waste into bis-hydroxyalkyl ethers of bisphenol A for use as PU raw materials†
Chao-Hsing
Lin
,
Hsing-Yo
Lin
,
Wei-Zhi
Liao
and
Shenghong A.
Dai
*
Department of Chemical Engineering, National Chung Hsing University, Taichung, Taiwan. E-mail: shdai@dragon.nchu.edu.tw; Fax: +886-4-2287-4159; Tel: +886-4-2285-1283
First published on 26th September 2006
Abstract
An efficient process for the chemical recycling of polycarbonate (PC) waste into diols of bis-phenol A (BPA) for use as raw materials in PU production has been developed. Digestion of PC waste in alkylene glycols, i.e., ethylene glycol (EG) and propylene glycol (PG), with a catalytic amount of sodium carbonate at 180 °C under normal atmospheric pressure afforded partial alkoxylation products of BPA. This initial product mixture was found to consist of BPA (28%) and monohydroxyethyl ether (MHE-BPA, 40%) and bishydroxyethyl ether of bisphenol A (BHE-BPA, 25%) when digested in EG solution. Whereas, in PG digestion solution, the corresponding digestion products consist of monohydroxypropyl ether of BPA (MHP-BPA, 53%) and bishydroxypropyl ether of BPA (BHP-BPA, 21%), with the rest being un-propoxylated BPA. When these digested solutions were further treated with molar excess of urea with a catalytic amount of zinc oxide at 180 °C, BHE from EG or BHP-BPA from PG was produced in high yields. In both digestion processes, cyclic alkylene carbonates, i.e.ethylene carbonate (EC) or propylene carbonate (PPC), were observed as the transient intermediates which eventually disappear during the prolonged alkoxylation conditions. ZnO was added to the second step as the catalyst to accelerate urea's trans-esterification process. Both BHE and BHP products have been sucessfully utilized in making PU polymers with good mechanical properties. This chemical approach thus provides a viable alternative for PC recycling.
Introduction
Since the 1950s, polycarbonate (PC) has been successfully developed and used as a commodity engineering thermoplastic because of its high-performance properties, such as high transparency, temperature resistance and toughness. More recently, the market consumption of PC has increased even more rapidly due to penetration of its applications into the electronic and media industries. However, PC's commercial success has been marred partially by the problems of its waste at both the post-manufacturing and post-consumer stages. It was estimated that more than 6000 billion discs of PC waste were generated worldwide in 2005 alone which amounted to more than 13 million pounds in one year.1 In a time of growing concern over environmental protection, reuse of resources has become an important issue for PC also. In addition, the demand for material reuse has become more acute nowadays because of the sharp increase in oil prices. PC plastics were known to be recyclable by means of re-grinding followed by re-extrusion, but the quality of the PC suffered and it generally could not be used in high performance applications.2 Retro-polymerization or digestion of PC waste into monomeric compounds and reuse in polymer synthesis as raw material has also been touted as another solution to the PC waste problem. In this way, potentially more avenues of applications could be developed.One recent approach to the chemical recycling of PC wastes investigated by Oku and his co-workers was to digest them back into BPA and hydroxyethyl ethers of BPA, by treating the PC with ethylene glycol using NaOH (0.1 equiv.) as the promoter.3,4 In the digestion step, fast de-carbonylation of PC occurred with formation of partial alkoxylation products of BPA. The digestion ended up with formation of a product mixture consisting of BPA, monohydroxyethyl ether of BPA and bishydroxyethyl ether of BPA. In the second step of their process, the digested mixtures were further treated with additional pure ethylene carbonate (EC) to form bishydroxyethyl ether of BPA as the final product. This diol can then be used as a raw material in polymer synthesis.
The industrial feasibility of Oku's process seems to very much hinge upon the availability of EC which is not yet a commodity product despite many inexpensive routes to EC being developed recently. For instance, EC can be made from the reaction of ethylene oxide (EO) and carbon dioxide.5 In the 1990s, a newer process for the production of alkylene carbonates such as EC from the reaction of the corresponding alkylene glycols and urea, in the presence of metal oxide catalysts6,7 became known. The latter process can proceed at atmospheric pressure and at a temperature from 130 to 200 °C. This process seems to be safer and more applicable than the pressurized reaction required for the EO and carbon dioxide route.
In view of these studies, we have further advanced Oku's original process by developing a more convenient one-pot atmospheric process of alkoxylation.3 In the process, the first digestion step is similar to Oku's first digestion step. However, we have adopted the inexpensive urea as our carbonylation agent in glycols in the subsequent step. By doing so, cyclic carbonates were generated in situ from ethylene glycol or propylene glycol and were utilized to complete the ethoxylation or propoxylation process in formation of diols of BPA as the final products. We have demonstrated that bisalkoxylated diols of BPA can be formed in one-pot and then be reused in polyurethane as raw materials to form high molecular weight elastomers and plastics.
Experimental
Materials and reagents
Poly-bisphenol A carbonate, PC (BPA; analytical grade, Aldrich), waste PC was spent PC from the injection process using LG-Dow (medical grade), bisphenol A (polycarbonate grade, Showa), ethylene glycol (analytical grade, Aldrich), propylene glycol (analytical grade, ARCOS), ethylene carbonate (EC; analytical grade, Lancaster), propylene carbonate (PPC; analytical grade, ARCOS), 4,4′-methylene-bis(phenyl isocyanate) (MDI; analytical grade, Bayer), sodium carbonate (analytical grade, TEDIA), zinc oxide (analytical grade, TEDIA), polytetramethylene glycol, PTMEG (commercial product Terathane 2000, Dupont).Instrumentation
Infrared (IR) spectra were recorded with a Perkin Elmer Spectrum One FT-IR spectrometer and the testing samples were cast as potassium bromide pellets. High-performance liquid chromatography (HPLC ) spectra were run on a Shimadzu LC-6A using a HYPERSIL-100 C18 column, Thermo Electron Corporation, with a length of 250 mm, and i.d. 4.6 mm through a UV detector with a 254 nm light source. 1H-NMR spectra were recorded on a Varian Unity Inova FT-NMR (200 MHz) spectrometer. GPCs were run on a Waters Apparatus 515 HPLC pump 717 Autosampler, using a 2410 Refractive Index Detector. Thermal gravimetric analysis (TGA) was performed on a Seiko SII SSC/5200 at a heating rate of 10 °C min–1. Differential scanning calorimetry (DSC ) was performed on a Seiko SII SSC/5200 at a heating/cooling rate of 10 °C min–1. Gas chromatograph (GC ) spectra were run on a Finnigan 9001 with a column of QUADREX 007 series methyl silicone (ionization detector, FID). Stress–strain measurements on polymer samples were performed on a Hung Ta HT-8504 tensile tester at an elongation rate of 100% min–1. Tensile bars were punched from a solution-cast film of the polymers. The tensile bars were prepared with the following dimensions: length = 22 mm, width = 5.0 mm, and thickness = 0.50 mm. Hardness measurements were performed on Teclock Durometer GS-702N (Shore D) and GS-709N (Shore A) in compliance with ASTM D2240A.One-pot digestion and alkoxylation for converting PC into bis-hydroxyalkyl ether of BPA
| Process | Conversion of ethoxylation (%) | Yield (%) | Conversion of propoxylation (%) | Yield (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| BPA | MHE-BPA | BHE-BPA | Oligomers | BPA | MHP-BPA | BHP-BPA | Oligomers | ||||
| a Retro-polymerization. b Alkoxylation. c Not observed. | |||||||||||
| PC pellets | Ra | 28 | 40 | 25 | 2 | —c | 26 | 53 | 21 | 0 | — |
| Ab | 0 | 2 | 91 | 1 | 99 | 0 | 1 | 97 | 2 | 90 | |
| PC wastes | R | 27 | 40 | 23 | 0 | — | 23 | 54 | 23 | 0 | — |
| A | 0 | 4 | 84 | 5 | 92 | 0 | 1 | 95 | 3 | 98 | |
BHE-BPA. 1H-NMR (300 MHz, CDCl3)3δ 1.63 (s, 6H), 2.03 (t, J = 6.3 Hz, 2H), 3.91–3.96 (dt, J = 6.3, 4.5 Hz, 4H), 4.06 (t, J = 4.5 Hz, 4H), 6.80–7.15 (m, 8H).
BHP-BPA. 1H-NMR (200 MHz, CDCl3) δ 1.24–1.36 (m, 6H), 1.63 (s, 6H), 2.33 (s, 2H), 3.69–4.23 (m, 6H), 6.78–7.15 (m, 8H).
Synthesis of PU polymers with diols prepared
Results and discussion
Digestion of polycarbonate or PC waste by alkylene glycols in the presence of sodium carbonate
In our chemical recycling of polycarbonate (PC) into BPA-diols, two basic reactions, digestion and alkoxylation, were involved and were basically carried out sequentially in tandem. The first step of digestion involved the decarbonylation of PC with action of either ethylene glycol (EG) or propylene glycol (PG) at 180 °C. It was found that alkylene carbonates are generated during the digestion process in the presence of Na2CO3, but the cyclic carbonates were found to be quickly consumed to produce the partial alkoxylation products of BPA with concurrent evolution of carbon dioxide. Without addition of any carbonylation reagents at this stage, the digestion product gave only approximately 50% of all phenolic alkoxylation products of BPA. This is because two phenolic groups were generated from PC in formation of only one cyclic carbonate.In the digestion step, the reaction could be best monitored by the disappearing absorption band of the carbonyl group at 1785 cm–1, Fig. 1 (a), of PC in its IR spectroscopy to signify the complete conversion of PC into alkoxylated products. At 180 °C, this digestion step generally took about 20–30 minutes. The mixtures obtained after digestion with ethylene glycol (EG) were analyzed by HPLC and found to consist of bisphenol A (BPA, 28%), monohydroxyethyl ether of BPA (MHE-BPA, 40%) and bishydroxyethyl ether of BPA (BHE-BPA, 25%). Some intermediate products were also detected in the mixture.
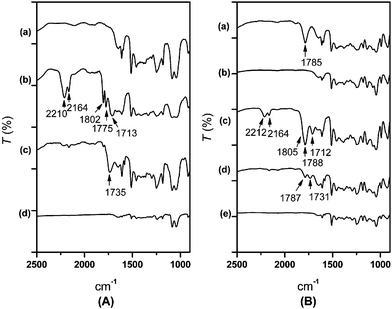 | ||
| Fig. 1 IR spectra of PC–EG digestion monitored. (A): (a) Digestion of PC by EG at 180 °C for 20 min, (b) further ethoxylation (urea) at 180 °C for 5 min, (c) 70 min, (d) 90 min. IR spectra of PC–PG reaction monitored. (B): (a) Digestion of PC by PG at 180 °C for 10 min, (b) digestion for 40 min, (c) further propoxylation (urea) at 180 °C for 10 min, (d) 80 min, (e) 120 min. | ||
The digestion of PC with propylene glycol (PG) gave three similar major compounds which were analyzed to consist of bisphenol A (BPA, 26%), monohydroxypropyl ether of BPA (MHP-BPA, 53%) and bishydroxypropyl ether of BPA (BHP-BPA, 21%). PC waste digestion by EG or PG gave consistently similar results to that of pure PC as shown in Table 1. The conversions were measured by HPLC .
Complete alkoxylations of phenolic in digested mixtures into bishydroxyalkyl ethers of BPA were accomplished by addition of extra carbonylation reagent, urea (1.8–2.0 equiv.) and ZnO (0.05–0.1 equiv.) as a catalyst. In the presence of glycols, urea generated more cyclic alkylene carbonates for alkoxylation. The cyclic alkylene carbonates reacted with BPA and mono-phenolic BPA moiety in the digestion products to produce bishydroxyalkyl ethers of BPA as shown in the reactions indicated in Scheme 1 (Eq. (1) and (2)).
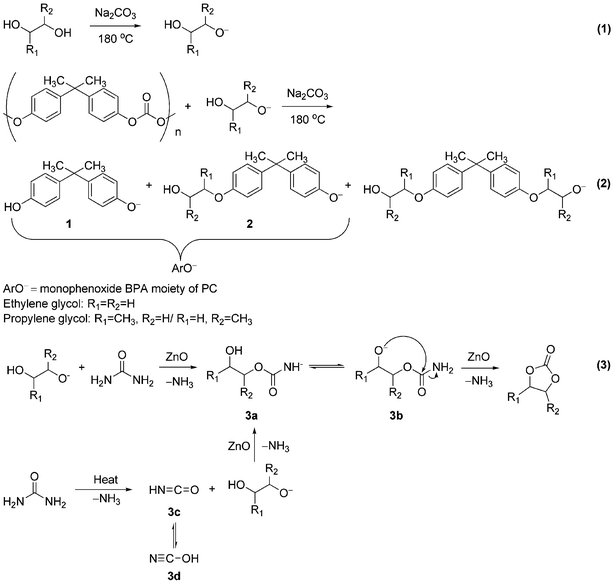 | ||
| Scheme 1 Conversion of PC into bishydroxyalkyl ether of BPA with alkylene glycol in the presence of urea. | ||
One-pot alkoxylation by urea–glycol in the presence of zinc oxide
In the alkoxylation of BPA's digested products, urea catalyzed by trace ZnO was used as the key carbonylation reagent. Urea would react with the alkylene glycols to generate cyclic alkylene carbonates initially to supplement further alkoxylation. Essentially, this is an extension of Doya's9 chemistry to BPA digestion products without the isolation of pure cyclic carbonates. In our one-pot approach to prepare BPA based diols, this step was generally carried out after the digestion of PC. In the urea–EG case, absorption characteristic of ethylene carbonate (EC) formation could be observed at 1802 cm–1 (Fig. 1 (A) (b)) at the early stage of the reaction. The absorption band characteristic of formation of propylene carbonate (PPC) was observed at 1785 cm–1 by IR (Fig. 1 (B) (c)). Both bands would disappear as the reaction proceeded to completion to form the final BPA diols.Mechanistically, the formation of cyclic alkylene carbonates from urea and glycols could result from two different pathways as indicated in Scheme 1. One pathway may proceed through attack of glycols at the carbonyl carbon of urea with elimination of NH3 to produce intermediate 3a (Scheme 1, Eq. (3)). The other pathway is directly from thermal decomposition10 of urea to produce NH3 and isocyanic acid (3c, HNCO)11,12 as the first step. Spectral evidence from our reaction solutions indicated that both pathways seemed operative. The characteristic absorption bands for HNCO could be observed at 2210/2212 cm–1 and its isomer HOCN (3d) is also detected at 2164 cm–1 by IR spectroscopy (Fig. 1 (A) (b)/(B) (c)).13 Therefore, the addition of alkylene glycols to HNCO is shown to be another route in the production of adduct 3a. Finally, adduct 3b would go through an intramolecular cyclic reaction and eliminate another mole of NH3 to produce cyclic alkylene carbonates. In short, BPA alkoxylated diols eventually became the final products of our one-pot process through cyclic carbonates' alkoxylation.
Ethoxylation of BPA with ethylene carbonate under the influence of sodium carbonate is very similar to ethoxylation of BPA with ethylene oxide (EO). In both cases BHE-BPA will become the major product.14 However, in most of our experiments, only small amounts of oligomeric polyether diol (1%) were detected as impurities in urea–EG ethoxylation. This extended reaction appeared to occur in the case when excess of urea and prolonged reaction time were applied. However, when an appropriate amount of urea was added, a high yield of BHE-BPA (96%) could be achieved. Thus, our results indicate that alkoxylation with cyclic carbonate results in more selectivity than EO for synthesis of mono-ethoxylated products from phenols when the catalyst used is sodium carbonate.
Selectivity and steric hindrance effect in propoxylation with urea–PG mixture
Similar to propoxylation with propylene oxide (PO), we have found three major propoxylated diols of BPA14 in the digestion products, i.e., primary–primary (1°-, 1°-), primary–secondary (1°-, 2°-), and secondary–secondary (2°-, 2°-) in a relative conversion ratio of 2 : 24 : 70, using our urea–PG one-pot process. These products of propoxylation resulted from the attack of phenate ions of BPA on the unsymmetrical structure of propylene carbonate. When the reaction takes place by route (1), attack on a less-hindered carbon of PC (Scheme 2), it produces products with the secondary hydroxyl group as the major product. In contrast, when ArO– attacks the alkylene carbon (C2) with a methyl group via more hindered route (2), the alcohol with a primary hydroxyl group becomes the product. The selectivity of our propoxylation does show a strong steric effect by showing high selectivity of 70–74% for 2°-, 2°-diols as the primary product and a low percentage (less than 2%) of 1°-, 1°-BPA diol in the propoxylated products. In addition, oligomers formation in the PG–urea reaction catalyzed by sodium carbonate was even lower than in the EG–urea reaction. Therefore, this one-pot synthesis is especially selective for preparing a mono-propylene ether extension for phenols.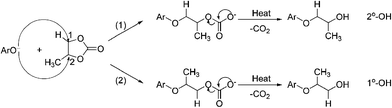 | ||
| Scheme 2 Diversity of products formation from propoxylation. | ||
Synthesis of PU with bis-hydroxyalkyl ether of bisphenol A and MDI
Among many potential applications of bis-hydroxyalkyl ethers of bisphenol A, utilization of them as raw materials in production of polyurethane (PU) seems to be one of the most direct. In our study, two types of PUs, plastic and elastomer, were prepared using BHE-BPA and BHP-BPA. For the synthesis of PU plastic, BHE-BPA and BHP-BPA were reacted individually with methylene diphenylene diisocyanate (MDI) in DMF and the products are designated as PU3 and PU4 respectively in Table 2. BHE-BPA and BHP-BPA were also used as the chain-extenders and were reacted with PTMEG-MDI pre-polymer in syntheses of PU elastomers to form PU1 and PU2 respectively. The molecular weight of PU1–4 was measured by means of GPC with DMF as solvent as shown in Table 2. The data indicate that both PU3 and PU4 are high polymers with very limited solubility in tetrahydrofuran solution.| Polymer | Compositions (molar ratio) | Hardness/shore | T d a /°C | T g,h b /°C | 100% Young's modulus /MPa | Strength/MPa | Strain at break (%) | Mw/g mol–1 | PDI |
|---|---|---|---|---|---|---|---|---|---|
| a Determined at a heating rate of 10 °C min–1. b h = hard segment. c Not observed. | |||||||||
| PU1 | MDI/BHE-BPA/PTMEG 2000 (1/0.8/0.2) | D 43 | 291 | —c | — | 11 | 48 | 27590 | 1.8 |
| PU2 | MDI/BHP-BPA/PTMEG 2000 (1/0.8/0.2) | A 86 | 289 | — | 6.6 | 15 | 341 | 64959 | 3.1 |
| PU3 | MDI/BHE-BPA (1/1) | — | 285 | 81 | — | — | — | 188106 | 1.6 |
| PU4 | MDI/BHP-BPA (1/1) | — | 279 | 123 | — | — | — | 45870 | 1.8 |
During polymerization, we found that the reactivity of BHE-BPA is far higher than that of BHP-BPA. This phenomenon is consistent with the fact that BHE-BPA is made entirely of primary hydroxyl groups. This could also explain why the molecular weight of PU4 was found to be only half that of PU3. However, both diols afforded PU of unique mechanical properties as illustrated in the observation below.
Thermal and mechanical properties of PU prepared
Thermal behavior of PU1 and PU2 was measured by means of TGA and DSC . The temperatures of 5% mass loss for PU1 and PU2 were 291 and 289 °C respectively in nitrogen as determined by TGA. DSC was used to obtain characteristics of the thermal transitions of PU1 and PU2. The first cooling and second heating traces of PU1 and PU2 are shown in Fig. 2 (A) and (B). The compositions of PU3 and PU4 consist of MDI with BHE-BPA or with BHP-BPA without polyols. The Tg,h of PU3 and PU4 were observed at 81 and 123 °C respectively (Fig. 3).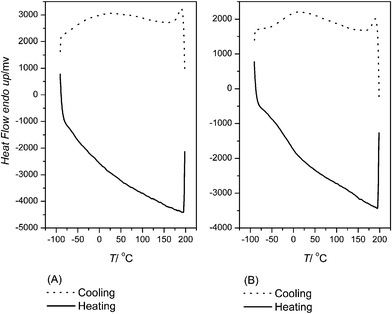 | ||
| Fig. 2 DSC traces of PU1 (A) and PU2 (B) at a heating/cooling rate of 10 °C min–1. | ||
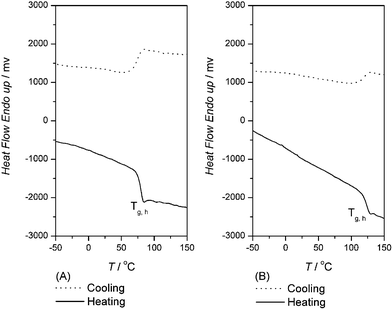 | ||
| Fig. 3 DSC traces of PU3 (A) and PU4 (B) at a heating/cooling rate of 10 °C min–1. | ||
For PU1 and PU2, no signs of melting nor crystallization peaks for the hard and soft segments could be discerned. This implies that both PUs are completely amorphous with a phase mixed between hard and soft phases. Chain extenders BHE-BPA and BHP-BPA both have two methyl groups on the methylene bridge in their molecular structures that are different from that of MDI. The steric hindrance caused by dimethyl group substitutions on the methylene bridges of the diols appears to block the tight packing of benzene rings and in turn causes the hard segment of PU in a random orientation. The phenomenon of thermal behavior from DSC seems consistent with these observations.
Films of PU1 and PU2 were cast from their DMF solutions, and this procedure produced completely transparent, flexible films. Hardnesses of PU1 and PU2 are measured to be D43 and A86 as shown in Table 2. Tensile bars of all PUs were punched from these non-oriented films and measured for their tensile strengths. Table 2 shows the stress–strain behavior of the two materials. Strain at break is expressed as the elongations, in terms of percentage. In PU1, 100% Young's modulus can not be measured, but the data shown for PU2 are totally different. The same formulation but different chain extenders exhibited completely different properties. PU1 behaved as a plastic but PU2 behaved as an elastomer. Again, the most logical reason is the branched methyl groups on the structure of the hydroxyethyl ether group in MHP-BPA. The branched methyl groups not only increase the solubility of bishydroxyalkyl ether of BPA (MHE-BPA is solid and MHP-BPA is liquid at room temperature), but also obstruct the intermolecular packing of the polymer. This result is reflected consistently in the enhanced hardness of PU1 that is D type.
Conclusions
Both pellets and wastes were converted into the hydroxyalkyl ethers of bisphenol A for use in polyurethane production. One-pot reactions with PC digestion and alkoxylation of the digested product with urea were developed. The yields of the BPA based diol were accomplished in excess of 90%. The selectivities of these urea-based alkoxylations were analyzed completely. A key use of the alkoxylation is the formation of cyclic alkylene carbonates with alkylene glycols and additional urea from an outside source in the presence of ZnO. This is a unique and economic route to produce different alkoxylation diols of BPA from PC wastes. They can be used as raw materials in the preparation of PU by a one-shot process. We have shown that PU possessing unique mechanical properties can be prepared from these recycled diols. We feel that PU3, PU4 and PU1 appear to possess properties which might make them strong candidates to replace PVC in the market. The detailed information on further endeavor on this green technology along this line of development is forthcoming.Acknowledgements
Financial assistance from Kuo-Ching Company of Taichung and from the Green Chemistry Team of the Center for Advanced Industry Technology and Precision Processing, NCHU of Taichung, Taiwan is gratefully acknowledged.References
- W. Y. Chen, Master's Thesis, Chaoyang University of Technology, 2005 Search PubMed.
-
G. Witt, US Pat., 5
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 619898, 1997 Search PubMed;
H.-J. Buysch, US Pat., 5652275, 1997 Search PubMed.
619898, 1997 Search PubMed;
H.-J. Buysch, US Pat., 5652275, 1997 Search PubMed. - A. Oku, S. Tanaka and S. Hata, Polymer, 2000, 41, 6749–6753 CrossRef CAS.
- L. C. Hu, A. Oku and E. Yamada, Polymer, 1998, 39, 3841–3845 CrossRef CAS.
-
J. T. Dunn and C. W. Va, US Pat., 2773881, 1956 Search PubMed;
C. H. McMullen, J. R. Nelson, B. C. Ream and J. A. Sims, Jr., US Pat., 4314945, 1982 Search PubMed.
-
W. Y. Su and G. P. Speranza, US Pat., 5003084, 1991 Search PubMed.
-
M. Doya, Y. Kanbara, K. Kimizuka and T. Okawa, US Pat., 5440004, 1995 Search PubMed.
-
R. Michel and L. Jacques, Fr. Pat., 1366067, 1964 Search PubMed.
-
M. Doya, T. Ohkawa, Y. Kanbara, A. Okamoto and K. Kimizuka, US Pat., 5349077, 1994 Search PubMed.
- B. Mavis and M. A. Kinc, J. Am. Ceram. Soc., 2006, 89, 471–477 CrossRef CAS.
- O. Krocher, M. Elsener and M. Koebel, Anal. Chim. Acta, 2005, 537, 393–400 CrossRef.
- M. U. Alzueta, R. Bilbao, A. Millera, M. Oliva and J. C. Ibanez, Energy Fuels, 2000, 14, 509–510 CrossRef CAS.
- C. Wentrup and H. Bornemann, Eur. J. Org. Chem., 2005, 4521–4524 CrossRef CAS.
-
S. Nobuyuki and T. Masahide, Jap. Pat., 2000128979, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: Conversion of (A) BHE-BPA and (B) BHP-BPA; Fig. S2: Selectivity of (A) BHE-BPA and (B) BHP-BPA. See DOI: 10.1039/b609638g |
| This journal is © The Royal Society of Chemistry 2007 |