Synthesis and applications of superacids. 1,1,2,2-Tetrafluoroethanesulfonic acid, supported on silica
Mark A.
Harmer
*a,
Christopher
Junk
a,
Vsevolod
Rostovtsev
a,
Liane G.
Carcani
a,
Jemma
Vickery
b and
Zoe
Schnepp
b
aDuPont Central Research and Development, Experimental Station, Wilmington, Delaware, 19880, USA. E-mail: Mark.A.Harmer@USA.DuPont.Com
bSchool of Chemistry, University of Bristol, UK
First published on 2nd October 2006
Abstract
In this paper we focus on the synthesis and use of superacids, in particular 1,1,2,2-tetrafluoroethanesulfonic acid(TFESA), and describe how these can be optimized for reactions of key industrial importance. One area of considerable interest is the field of superacid catalysis and, specifically, the development of safer and more cost-effective acid catalysts. We report a new simplified route for preparation of these acids, making these more readily available and opening up a large number of opportunities. Partially fluorinated superacids offer several advantages over the acids commonly used in catalysis (sulfuric, hydrofluoric acid and aluminium chloride): lower loadings, lower reaction temperatures (leading to increased selectivity), fewer by-products, shorter reaction times and higher throughput. TFESA and its longer chain analogs are much less volatile than triflic acid (CF3SO3H). We tested these superacids in several processes (aromatic alkylation, acylation of arenes, isomerization, oligomerization and the Fries rearrangement). These materials are excellent acid catalysts, comparable to triflic acid, and yet easier to handle. We have also prepared supported versions of these catalysts and introduced the ability to recycle.
Introduction
Sustainable growth is critical for both highly developed and developing countries. The term ‘sustainable growth’ is best defined as a way to meet societal needs while reducing our environmental footprint. From an industrial point of view this offers a wide variety of opportunities. These include the development of materials from renewable resources (such as alcohols from corn, and the production of bio-derived polymers), the optimization of industrial processes by catalysis (such as replacing homogeneous catalysts with heterogeneous ones), improvements in atom economy, development of renewable sources of energy, optimization of energy conversion using new technologies such as fuel cells and the continued development of hybrid car engines.1In this paper we focus on the use of Brønsted superacidic catalysts and describe how these can be utilized for a number of acid catalyzed reactions of industrial importance. We also show that supported versions of these acids show superior properties to the free acid. Traditional homogeneous acids such as hydrofluoric(exceptionally hazardous) and sulfuric acids (often has be used in large volumes relative to the reactants) have a number of drawbacks in industrial processes and, furthermore, generate large amounts of waste associated with the separation of the product. Replacing these acids with highly active catalysts (such as superacids) that can be recycled and easily separated reduces waste, reduces energy needs, cuts down the process time and makes the overall process both safer and more economical. Superacidic materials are finding uses in an increasingly wide range of industrial applications. For example, triflic acid is already employed in fine chemical synthesis, batteries, and ionic liquids (anions). Interest in these materials is growing.2–4
A useful starting point in discussion of superacidic materials is the subject of acid strength. Superacids are defined as having a Hammett acidity constant (H0) of greater than –12. In essence, they are acids that are stronger than 100% H2SO4.5 It is exactly this property that gives these materials many advantages over traditional acids such as HF. The higher acid strength means that smaller amounts of acid can often be used. These superacids can also catalyze reactions at temperatures that are lower than those of sulfuric acid-catalyzed processes. In industrial catalysis, this can mean the difference between handling vast quantities of H2SO4, with all the inherent hazards it entails, compared to a small amount of a highly active alternative. Sulfuric acid is also inherently quite reactive which can, in turn, lead to unwanted by-products. These include sulfonate esters (reactions with alcohols) and also sulfonation of aromatic rings (arising from the oxidizing ability of sulfuric). Sulfate esters have to be removed by additional processing steps that lead to poorer yields (lower atom efficiency), additional work up costs and more waste. Triflic acid and the acids described in this paper do not suffer from the same draw backs. The anion, TFES, is non-coordinating and the formation of sulfate type esters is not a problem.
This paper focuses on the largely unused classes of superacids of the form RCHFCF2SO3H (R = F, Cl or fluoroalkyl). Although our research has included the potential of these materials for various applications, the main focus has been the use of these acids for catalysis. We also emphasize three aspects: the safe delivery of superacids, the effective use of superacids, and acid recycling.
The importance of acid catalysts in industry cannot be overstated. Acid catalysts, are employed in an extensive range of commercial reactions: alkylations, acylations, rearrangements and oligomerizations. Alkylations are common in the commercial production of ethyl benzene, trimethylpentane for high octane fuels, cumene and linear alkyl benzenes for the surfactants industry. Traditionally, homogeneous acid catalysts such as AlCl3, FeCl3, H2SO4, HF and BF3, are employed.6,7 Isomerization and olefin oligomerization are used in refining and preparation of lubricants.8 Acylation, a key reaction for the fine chemicals industry, currently utilizes AlCl3 or catalytic quantities of hazardous HF.9–11 Fries reaction is used in the production of hydroxyacetophenone, an important precursor for the formation of the painkiller p-hydroxyacetanilide, and is promoted by BF3.11
There are several problems associated with the traditional acid catalysts described above. The hazards of hydrofluoric acid pose severe dangers to people and the environment during both transportation and usage. Lewis acids such as FeCl3 and AlCl3 often need to be used in stoichiometric quantities, and subsequently create sizable waste streams. The current drive to use solid acid catalysts solves some of these issues, decreasing waste generation.11–13 As a result, recyclable materials and reduced waste lead to a more economical processes. However, it is very likely that both liquid and solid acids will be used in the foreseeable future.
Here, we describe the preparation and properties of fluoroalkylsulfonic acids such as 1,1,2,2-tetrafluoroethanesulfonic (TFESA) and 1,1,2,3,3,3-hexafluoropropanesulfonic (HFPSA) acids. We expect the acidity functions of these acids to be similar to triflic acid (CF3SO3H). As we discuss below, the catalytic conversions for a range of reactions are comparable to triflic acid. This is usually a reasonable guide for acid strength. The acidity function of CF3SO3H for example is similar to CF3CF2SO3H (H0 of –14.1 and –14 respectively). It is also known that appending a sulfonic acid group to either a CF2 or CF2 group such as CF2CF2 does not alter the acidity significantly.14 A range of applications of these acids as well as supported versions of TFESA will be described.
Results and discussion
Triflic acid is a well-known and commercially available superacid.2,11 In this paper we describe the synthesis of three alternate superacids (1–3) and a supported version of one of them (TFESA, 1). This paper will focus mainly on 1,1,2,2- tetrafluoroethanesulfonic acid (1). Compared to triflic, these acids have lower volatility which translates to ease of handling.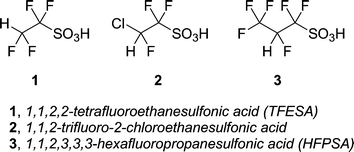
The boiling point of 1 (210 °C) is higher than that of triflic (165 °C) or hydrofluoric (19 °C) acid. Vapour pressures of the two acids are compared in Fig. 1. The lower vapour pressure of TFESA makes handling of the acid easier, improving safety. The higher boiling point also increases the processing window during which these acids may be used. A simple illustration showing the reduced volatility is shown in Fig. 2.
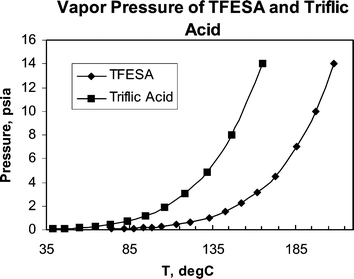 | ||
| Fig. 1 Vapour pressures of TFESA and triflic acid. | ||
 | ||
| Fig. 2 Visual comparison of triflic acid (left vial, 1 g) to TFESA (right vial, 1 g) in air after 1 min. | ||
In addition, all compounds under discovery in this article contain a hydrogen on the second carbon which acts as a spectroscopic fingerprint in the proton NMR spectrum, Fig. 3. This feature is especially useful in several ways, one of which is determining any potential contamination of the product with the acid. In biphasic type reactions NMR can also be used to determine distribution coefficients of the acid.
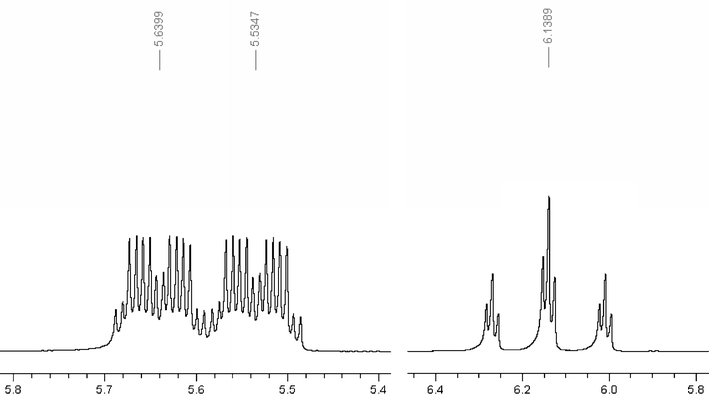
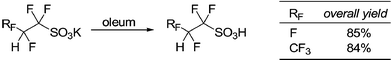
In terms of the synthesis, TFESA was first synthesized by Barrick.15 Subsequently, we have made a number of improvements in the synthesis.16 The literature describe the use of free radical initiators to facilitate the addition of the sulfite ion to fluorinated double bonds. We found that the reaction works well in the absence of the initiators and that it most likely occurs through a nucleophilic attack by the sulfite. The fluorinated carbanion, which is formed after sulfite addition, is protonated by water. In the absence of a buffer, the result is an increasingly basic solution that promotes hydrolysis of tetrafluoroethylene to the unwanted by-product difluoroacetate (HCF2COOM), as well as the desired product. Acidity of the reaction mixture can be conveniently controlled by a buffer solution comprised of sulfite and bisulfite ions. At pH 5–6, fluoroalkylsulfonates were obtained in high yields with little or no difluoroacetate impurities. Careful attention to pH (to prevent formation of the difluoroacetate) leads to better product selectivity (typically >99%).
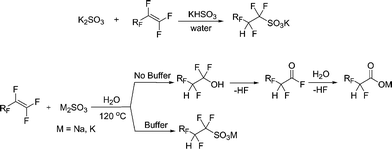
We have also found that sulfite addition to TFE can also be achieved by using TFE/CO2 mixtures. In this case the carbon dioxide acts as an internal buffer, again leading to greater selectivity.
Conversion of dried sulfonates to the corresponding acid is accomplished by distillation from oleum in one step. Alternatively, a two-step procedure can be used. First, acid hydrate is distilled from concentrated sulfuric acid. In the second step, it is dehydrated in thionyl chloride and redistilled again to afford anhydrous superacids. In summary, sulfite addition to TFE proceeds smoothly, with high selectivity, and high yield.
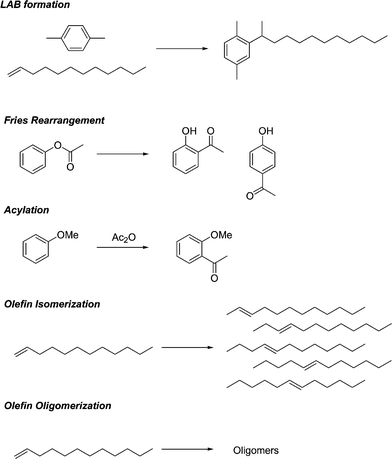
We investigated a number of reactions of industrial importance using the liquid superacids. These include the formation of linear alkyl benzenes (LAB), acylation, the Fries reaction, olefin isomerization and olefin oligomerization (Scheme 1).
 | ||
| Scheme 1 | ||
High catalytic conversions were obtained demonstrating the utility of these Brønsted acids, except in the case of olefin isomerization and oligomerization , where the immiscibility of these polar acids with the reaction mixture diminished the catalyst's activity. The results are shown below in Table 1.
| Catalyst type | Acylationa | Fries Reactionb | Alkylationc |
|---|---|---|---|
| a 100 mmol anisole, 100 mmol acetic anhydride at 1 h, 100 °C, 0.5 g of catalyst. b 25 g of phenylacetate after 4 h at 140 °C, 0.5 g of catalyst. c aromatic alkylation, 15 g of p-xylene and 5 g of 1-dodecene after 1 h at 100 °C, 0.5 g of superacid catalyst. | |||
| HCF2CF2SO3H | 63 | 71 | 99 |
| CF3HCFCF2SO3H | 59 | 69 | 98 |
| HClCFCF2SO3H | 58 | 49 | 95 |
| CF3SO3H | 61 | 58 | 98 |
| H2SO4 | 10 | — | <5 |
In alkylation reactions all of the superacids tested show quite high catalytic conversion and in all cases higher conversion was found compared to sulfuric acid. The formation of linear alkyl benzenes has been recently reviewed.6,7
These materials, when sulfonated, represent the basis of the detergents industry. In the current commercial process, HF is used as the catalyst and there is an obvious drive to replace this hazardous material. The products of these reactions contain a mixture of alkylbenzenes with the phenyl group attached to different carbon atoms in the linear hydrocarbon chain. The 2-phenyl isomer is the most preferred product. Branched isomers, which are a result of a skeletal isomerization of the linear hydrocarbon chain, are very undesirable due to lower biodegradability. For all of the superacids tested, catalytic conversions of 95–99% were obtained at 100 °C using xylene as the aromatic. The products contain >95% LABs and the rest (<5%) are the branched alkylates derived from the ∼4% branched olefins present in the feed, dimers of 1-dodecene and disubstituted benzene. The linearity of the alkylation using 1-dodecene is >99%. Sulfuric acid at a similar loading showed very low activity. Much higher loadings (approx. 20–30 wt% sulfuric) were needed to obtain comparable conversions.
Superacids are also finding increasing uses in fine chemicals synthesis.2 It is well known that the traditional catalysts used in acylation chemistry and the Fries reaction (used in the manufacture of an ibuprofen intermediate) suffer from several drawbacks, mainly the use of stoichiometric amounts of catalysts (AlCl3, BF3). This leads to extensive problems with the disposal of the associated waste streams. Again, the above superacids are all active in catalytic amounts and we believe the higher boiling superacids may have the additional advantage of ease of handling due to lower volatility. A number of papers have recently been published on the use of triflic acid and metal triflates, which have been recycled using ionic liquids as a reaction media.17,18 The ability to recycle is also important in terms of using these acids.
These acids were also investigated in the isomerization and oligomerization of 1-dodecene. The liquid superacids showed <1% conversion, essentially inactive. This can be explained by the immiscibility in the polar superacids and the non-polar reactant 1-dodecene. This problem was circumvented by developing solid supported versions of these acids. In comparison these show exceptionally high conversion for these same reactions.
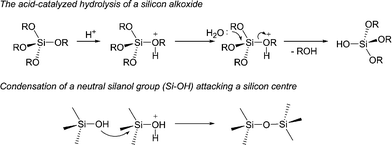
Silica supported superacids were prepared using both in situ sol-gel method and infiltration into pre-formed porous silica supports. In the case of in situ sol-gel, tetramethoxysilane was pre-hydrolysed with an excess of water. TFESA was then added in amounts such that the final loading was between 5 and 75 wt%. Upon addition of the acid a clear homogeneous solution formed, which gelled upon drying in an oven overnight (in nitrogen atmosphere at 80 °C). This procedure gives a hard, glass-like composite with the superacid highly dispersed within and throughout the silica network. Porous particles a few mm in size result.

The first step of this process is a hydrolysis of the alkoxides. The mechanism for this hydrolysis (catalysed by a small amount of an acid such as HCl) is well known (below) and results in the formation of polysilicic acid (Si(OH)4, not shown).19 Condensation occurs either by the reaction of two Si-OH groups or reaction of SiOH with SiOR groups leading to the formation of an extended silica network. These two steps occur simultaneously, although in the case of acid-catalyzed reactions hydrolysis occurs rapidly (minutes) followed by slower condensation (hours) at higher temperatures (typically 80 °C) and during the drying stages.
The optimum microstructure (in terms of an acid catalyst) had an acid loading of about 15 to 25 wt% of acid, surface area of about 439 m2 g–1, pore volume 0.74 cc g–1 and an average pore diameter of about 67 Å. We compared a 5, 25 and 50 wt% containing composite (weight of HCF2CF2SO3H in silica, same moles of acid used in catalysis, in the 1-dodecene alkylation of p-xylene). The resulting conversion of the 1-dodecene had values of 50, 82 and 40% respectively (100 °C, 2 hr. reaction time , Table 2). The highest catalytic conversion, had an acid loading within the silica of 25 wt%. A more detailed description will be published separately showing the optimization of the microstructure (in terms of catalyst loading, pore size and surface area). We also found that, in terms of acid stability, the acid does not leach in non-polar solvents such as benzene, hexanes, olefins, toluene and xylenes.
| Catalyst type | Alkylation (%)a | Isomerization (%)b | Oligomerization (%)c | Acylation (%)d |
|---|---|---|---|---|
| a 15 g of p-xylene and 5 g of 1-dodecene after 1 h at 100 °C, 1 g of 25 wt% supported catalyst and 0.25 g of HCF2CF2SO3H. b 25 g of 1–dodecene after 30 min at 100° with 1 g of supported acid. c 25 g of 1–dodecene after 30 min at 120 °C with 1 g of supported acid. d 100 mmol anisole, 100 mmol acetic anhydride after 1 h at 100 °C with 1 g of supported acid. | ||||
| HCF2CF2SO3H | 10 | <2 | <2 | 65 |
| Silica/HCF2CF2SO3H | 82 | 99 | 90 | 68 |
The distribution of the acid in the silica can be inferred by examining pore volume, surface area and pore diameter of the TFESA/silica network compared to the network where the acid had been leached out (using ethanol). A large increase in pore volume with small increase in pore diameter is consistent with an acid, highly dispersed within and throughout the silica network. The resulting acid-leached silica network had a pore structure (in the case of a 25 wt% containing microstructure as described above) with a surface area of about 540 m2 g–1, pore volume of 0.93 cc g–1 and a pore diameter of about 70 Å. The higher pore volume is consistent with the removal of the acid and a small increase in pore diameter noted (with no evidence for large pockets of extra pores).
In addition to the in situ route, we also found silica/superacid composites could be made by infiltration of pre-formed supports (Fig. 4). The as-received support has a surface area of about 345 m2 g–1, pore volume 1.22 cc g–1 and a pore diameter of about 141 Å.
 | ||
| Fig. 4 Silica supported TFESA (@1.5 mm beads). | ||
This pre-formed spherical support was infiltrated with an diethylether containing TFESA solution. The ether was then removed leaving the silica/TFESA catalyst. Again, the optimum loading was 15–25 wt% of TFESA. Comparison of the silica-supported TFESA prepared by the two approaches revealed that both afforded highly active catalysts for a range of reactions. In non-polar media the TFESA/silica showed higher catalytic conversion in a number of reactions, compared to the TFESA acid.
The silica-supported acids were surprisingly stable in terms of volatility. For example, 1 g of the free acid (TFESA) completely vaporizes when heated to 150 °C under a vacuum (2 h), whereas 85% of the acid in the silica-support was retained (even after 18 h). The TFESA/silica composite was also more stable than the triflic acid/silica composite. A triflic acid containing silica at a similar loading retained about 40 wt% of the original acid after 18 h. From a safety and handling point of view, this feature (very low volatility within the silica) makes these materials especially attractive for use in a wide variety of applications. Another advantage of the increase in stability is catalyst drying. Brønsted acids typically pick up moisture and have to be dried. The TFESA/silica composite can be readily dried with only a small amount of acid loss. The exact nature of the interaction within the support is not known, however the reduced volatility is consistent with hydrogen bonding of the silanols to the acids.
The catalytic conversion of these silica-supported acids was exceptionally high for a number of reactions. Four different types of reactions were investigated to compare the free and the supported acid, (Table 2). In the case of a non-polar reaction mix, very little leaching of the acid occurs. These materials behave as solid acid catalysts (heterogeneous catalysts) with all of the expected advantages compared to homogeneous catalysts. Advantages include ease of product separation, high activity, and the ability to recycle the catalyst. From an industrial point of view, this results in less waste and, inevitably, better economics.
The silica-supported superacids show high conversion in the alkylayion of p-xylene with 1-dodecene. The conversion is higher than the homogeneous acid itself (Table 2). The product is easy to separate from the catalyst and the catalyst can be easily recycled. At 100 °C, the supported acid shows almost complete conversion after 15 min, in contrast to the liquid TFESA (10% conversion, same molar amounts of acid in each case), Fig. 5. In these studies we reduced the acid loading (essentially 0.25 g vs. 0.5 g of TFESA, see Table 1) to highlight differences. Both the in situ sol-gel-derived material and the infiltrated pre-formed material had about the same activity. The extent of leaching was low, with values of about 0.2 to 0.5%, respectively, for a number of alkylation reactions, with about 99.8% of the acid retained within the silica. In a typical study the reaction products were separated from the catalyst and the acid content was determined, both in the organic phase (alkylate phase) and in the silica containing acid phase (see Experimental). We carried out the analogous reaction using a triflic-supported catalyst. The activity was quite low (40% compared to 82% using the TFESA) due to loss of the volatile triflic acid during the supported catalyst preparation and drying stage. The silica-supported TFESA catalyst was recycled twice with a slight decrease in activity (conversions for the 1-dodecene alkylation of p-xylene at 1 h of 98, 97, and 94%, three recoveries).
Olefin isomerization was also investigated. The isomerization of long chain olefins is often carried out using large amounts of sulfuric acid, and the isomerized product is subsequently used as a component in lubricants. Olefin isomerization involving cis-/trans-transformation, or double bond migration , is relatively easy chemistry.20 However, many solid acid catalysts quickly deactivate. The olefin isomer products are useful in the formulation of well fluids such as drilling mud, useful for offshore drilling. An Amoco patent by Clarembeau and Steylaerts used Amberlyst®, Nafion® resin pellets and Nafion® resin/silica composites to catalyze the isomerization of terminal olefins with 14 to 20 carbon atoms.20 The catalysts described herein (impregnation of a pre-formed support) are simpler than the Nafion® resin-based systems and may become viable alternatives. Whereas the free acid showed virtually no catalytic activity in this reaction, the supported TFESA catalyst showed equilibrium conversion to the six isomers within about 15 min at 80 °C.
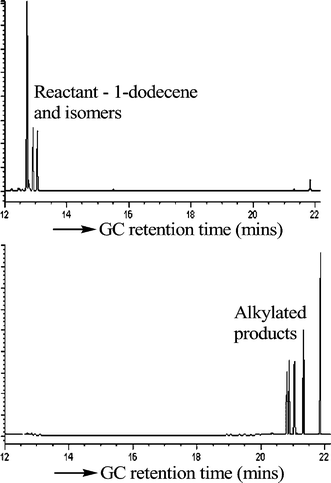 | ||
| Fig. 5 GC of the reaction of p-xylene (15 g, peaks not shown) with dodecene (5 g, GC retention time 13 min), after 15 min reaction time at 100 °C using liquid TFESA (0.25 g), top, and SiO2/TFESA (1 g, 25 wt% loading), bottom, showing higher activity of the supported superacid (y axis—a/units). Peaks of the alkylated product are found around 21 to 22 min. | ||
Oligomerization was also found to occur at higher temperatures (120 °C). 1-Dodecene is oligomerized to the dimers and trimers, again with high conversion (>95% of 1-dodecene, Fig. 6). Propyleneoligomerization to C6+ olefins, the famous UOP catalytic condensation process, catalyzed by solid phosphoric acid has been widely used in the petroleum industry for more than 70 years.12 The oligomerization is typically carried out in a fixed bed reactor at supercritical conditions, e.g. ∼200 °C and 1000 psig. The products are used as petroleum fuels. There is interest in finding replacements for the currently used catalyst, phosphoric acid, on silica. Both types of pre-formed catalysts (based on in situ and pre-formed silica) show high activity for dodeceneoligomerization . Therefore, these materials have tremendous potential for use in reactions of industrial importance.
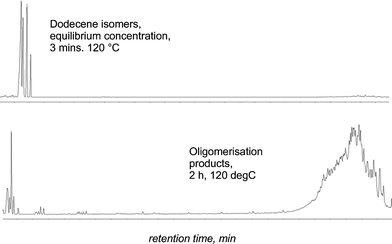 | ||
| Fig. 6 GC traces of the reaction of 1-dodecene (20 g) after 3 min (top) and 2 h (bottom) at 120 °C using TFESA/SiO2 (0.5 g each) showing rapid isomerization followed by oligomerization . (y axis—a/units). | ||
In summary, superacid catalysts based on fluoroalkylsulfonic acids can be produced via addition of sulfites to fluorinated olefins in buffered solutions. Catalysts of this type represent an important class of superacid catalysts. New, simplified and high-yielding routes to these compounds open up a wide variety of opportunities. The relatively low volatility and high boiling point translate to safer handling of these materials. Supported versions of these acids enable reactions in non-polar reaction media. The silica supported superacids described in this paper have been found to show high catalytic activity with low leaching in a number of reactions of industrial importance.
Experimental
Synthesis of TFESA
A 1-gallon Hastelloy C276 reaction vessel was charged with a solution of 175 g potassium sulfite hydrate (K2SO3·xH2O, 95%, Aldrich), 610g potassium metabisulfite (K2S2O5, 99%, Mallinckrodt) and 1500 mL of deionized water. The vessel was cooled to –35 °C, evacuated and then purged with nitrogen. The evacuate/purge cycle was repeated two more times. The vessel was heated to 120 °C. To the vessel was then added 500 g of tetrafluoroethylene via slow addition keeping the pressure below 200 psi. The total reaction time to complete the addition was about 5–6 hours. The reaction was allowed to cool to room temperature before venting excess gases and rinsing the reaction mixture from the shaker tube with deionised water. The water was removed in vacuum and the potassium salt was extracted with acetone. 19F NMR (D2O) δ: –122.0 (dt, 3JFH = 6 Hz, 3JFF = 6 Hz, 2F); –136 (dt, 2JFH = 53 Hz, 2F), consistent with tetrafluoroethanesulfonate (TFES-K). 1H NMR (D2O) δ 6.4 (tt, 2JFH = 53 Hz, 3JFH = 6 Hz, 1H). The acetone layer was removed, filtered, and then dried. This was then added to an excess of sulfuric and followed by addition of thionyl chloride to generate the dehydrated acid, which was purified by distillation (around 110 °C) on a vacuum line. Alternatively, a 100 mL round bottomed flask with a sidearm and equipped with a digital thermometer and magnetic stir bar was placed in an ice bath under positive nitrogen pressure. To the flask was added 45 g crude TFES-K (potassium and sodium salts can be used interchangeably), 30 g of concentrated sulfuric acid (95–98%) and 78 g oleum (20 wt% SO3) while stirring. The amount of oleum was chosen such that there would be a slight excess of SO3 after the SO3 reacted with and removed the water in the sulfuric acid and the crude TFES-K. The mixing caused a small exotherm, which was controlled by the ice bath. Once the exotherm was over, a distillation head with a water condenser was placed on the flask, and the flask was heated under nitrogen behind a safety shield. The pressure was slowly reduced using a PTFE membrane vacuum pump (Buchi V-500) in steps of 1.93 psi (100 Torr) in order to avoid foaming. A dry-ice trap was placed between the distillation apparatus and the pump to collect any excess SO3. When the pot temperature reached 120 °C and the pressure was held at 0.38 psi (20 Torr) a colorless liquid started to reflux which distilled at 110 °C and 0.57 psi (31 Torr). Proton NMR and analysis showed the product, TFESA, was greater than 99.5% pure with an overall yield of about 85%. Analysis gave the following results: 19F NMR (CD3OD) δ: –125.2 (dt, 3JFH = 6 Hz, 3JFF = 8Hz, 2F); –137.6 (dt, 2JFH = 53 Hz, 2F). 1H NMR (CD3OD) 6.3 (tt, 3JFH = 6 Hz, 2JFH = 53 Hz, 1H).Synthesis of 1,1,2,3,3,3-hexafluoropropanesulfonic acid(HFPSA):
A 1-litre Hastelloy® C 276 reaction vessel was charged with a solution of anhydrous sodium sulfite (25 g, 0.20 mol), sodium bisulfite 73 g, (0.70 mol) and deionized water (400 ml). The pH of this solution was 5.7. The vessel was cooled to 4 °C, evacuated and then charged with hexafluoropropene (HFP, 120 g, 0.8 mol). The vessel was heated with agitation (1000 rpm) to 120 °C and kept there for 3 hr. The pressure rose to a maximum of 335 psi and then dropped down to 32 psi within 30 minutes. At the end, the vessel was cooled and the remaining HFP was vented, and the reactor was purged with nitrogen. The final solution had a pH of 7.3. The water was removed in vacuo on a rotary evaporator to produce a wet solid. The solid was then placed in a vacuum oven 140 degrees C, 48 hr) to produce 219 g of white solid which contained approximately 1 wt% water. The theoretical mass of total solids was 217 g. The crude HFPS-Na can be further purified and isolated by extraction with reagent grade acetone, filtration, and drying. A 100 mL round bottomed flask with a sidearm and equipped with a digital thermometer and magnetic stir bar was placed in an ice bath under positive nitrogen pressure. To the flask was added 50 g crude sodium hexafluoropropanesulfonate (HFPS-Na), 30 g of concentrated sulfuric acid (95–98%) and 58.5 g oleum (20 wt% SO3) while stirring. The amount of oleum was chosen such that there would be a slight excess of SO3 after the SO3 reacted with and removed the water in the sulfuric acid and the crude HFPSA. The mixing caused a small exotherm, which was controlled by the ice bath. Once the exotherm was over, a distillation head with a water condenser was placed on the flask, and the flask was heated under nitrogen behind a safety shield. The pressure was slowly reduced using a PTFE membrane vacuum pump in steps of 1.93 psi ( 100 Torr) in order to avoid foaming. A dry-ice trap was placed between the distillation apparatus and the pump to collect any excess SO3. When the pot temperature reached 100 °C and the pressure was held at 0.38 psi (20 Torr) a colorless liquid started to reflux and later distilled at 118 °C and 0.57 psi (23 Torr). A forerun of lower-boiling impurity (1.5 g) was obtained before collecting 36.0 g of the desired acid, hexafluoropropanesulfonic acid (HFPSA). An 84% overall yield from HFP was obtained. 19F NMR (D2O) δ: –74.5 (m, 3F); –113.1, –120.4 (ABq, J = 264 Hz, 2F); –211.6 (m, 1F). 1H NMR (D2O): 5.8 (m, 2JFH = 43 Hz, 1H).Synthesis of 1,1,2-trifluoro-2-chloroethanesulfonic acid
The 1,1,2-trifluoro-2-chloroethanesulfonic acid was prepared according to the procedure described for HFPSA, but using 1,1,2-trifluoro-2-chloroethene instead of HFP. Obtained yields were ca. 80%.Synthesis of TFESA/Silica
Tetramethylorthosilicate (TMOS, 160 g), water (188 g) and HCl (0.1 M, 1 g) were stirred together for 20 minutes to hydrolyze the tetraalkoxide. HCF2CF2SO3H (20 g) was then added and the mixture left stirring for three hours in an open jar at room temperature. The resulting (still liquid) mixture was placed in a 80 °C oven for 2 days to gel and harden. Drying was completed in a 100 °C vacuum oven for 24 h. Pore diameter and volume were measured by BET method.Catalyst testing
All reagents were reagent grade. The catalyst (solid) was dried at 150 °C overnight before use under vacuum. Acylation: the acid catalyst was added to a dry round bottom flask under a nitrogen atmosphere and anisole 100 mmole and acetic anhydride (freshly distilled) 100 mmole were added. The sealed flask was transferred to a 100 °C oil bath and stirred vigorously. Samples were diluted 1 in 20 in ether for GC analysis. All of the samples were analyzed by a Hewlett Packard 5890 Series II GC equipped with FID detectors. Product identification was carried out with a GC-MS analysis and 1H NMR. The products were identified by comparison of their spectra and retention time in GC with those of authentic samples (for example para-acetophenone in the acylation reaction studied). In the case of the alkylation reactions (carried out at 100 C with conversion close to 99%), the products contain >95% linear alkylate and the remainder (<5%) are the 4% branched alkylates from the 4% branched olefins (impurity in the feed), and dimers of 1-dodecene. Full details for these reactions has been reported previously21–24 The Fries reaction, alkylation, oligomerization were all carried out in a similar manner analyzing conversion using gas chromatography. Acid leaching, in the case of 1–dodecene alkylation of p-xylene, was measured by first filtering the products from the solid supported acid. The organic mix was then extensively stirred with water to extract the acid (4 times the volume, >48 hrs), and the water containing layer was titrated. Additional water was added to ensure all the acid had been removed. The acid containing water layer was titrated against base to determine the acid content. The filtered solid, containing the bulk of the acid, was also titrated (again extensive water washing to remove the acid). A good materials balance was obtained with typically greater than 99.5% retention of the acid in the solid (0.5% in the organic product phase).Acknowledgements
We would like to thank the invaluable help of Bob Miller, Mike Barker, Keiiche Tanabe, Qun Sun, Ed Howard and Monica Tisack, for help on synthesis and many helpful technical discussions.References
- P. T. Anastas and T. C. Williamson, in Green Chemistry: Frontiers in Benign Chemical Syntheses and Processes, Oxford University Press, Oxford, 1998 Search PubMed.
- J.-P. Simonato, Chem. Ind., 2005, 20 CAS.
- E. Marx, Spec. Chem. Mag., 2004, 24, 24 Search PubMed.
- Y. Katsuhara, M. Aramaki, A. Ishii, T. Kume, C. Kawashima and S. Mitsumoto, J. Fluor. Chem., 2006, 127, 8 Search PubMed.
- G. A. Olah, G. K. Surya Prakash and J. Sommer, Superacids, Wiley-Interscience, New York, 1985 Search PubMed.
- A. de Angelis, C. Flego, P. Ingallina, L. Montanari, M. G. Clerici, C. Carati and C. Perego, Catal. Today, 2001, 65, 363 CrossRef CAS.
- J. A. Kocal, B. V. Vora and T. Imai, Appl. Catal., A, 2001, 221, 295 CrossRef CAS.
- S. J. Miller, Stud. Surf. Sci. Catal., 1994, 84, 2319 CAS.
- G. A. Olah, Interscience Monographs on Organic Chemistry: Friedel–Crafts Chemistry, Wiley-Interscience, New York, 1973 Search PubMed.
- J. A. Horsley, Chemtech, 1997, 27, 45 CAS.
- G. Sartori and R. Maggi, Chem. Rev., 2006, 106, 1077 CrossRef CAS.
- M. A. Harmer, in Handbook of Green Chemistry & Technology, ed. J. Clark and D. MacQuarrie, Blackwell Publishing, Oxford, 2002 Search PubMed.
- M. A. Harmer, W. E. Farneth and Q. Sun, Adv. Mater., 1998, 10, 1255 CrossRef CAS.
- G. A. Olah, in Acidity and Basicity of Solids, ed. J. Fraissard and L. Petrakis, Kluwer Academic, Boston, 1994, p. 305–334, Search PubMed.
- D. D. Coffman, M. S. Raasch, G. W. Rigby, P. L. Barrick and W. E. Hanford, J. Org. Chem., 1949, 14, 747 CrossRef CAS.
- M. A. Harmer, C. P. Junk and Z. Schnepp, US Pat. App., filed on 06/07/2005 Search PubMed.
- J. Ross and J. Xiao, Green Chem., 2002, 4, 129 RSC.
- V. D. Sarca and K. K. Laali, Green Chem., 2004, 6, 245 RSC.
- G. W. Scherer and C. J. Brinker, Sol-Gel Science, Academic, San Diego, 1990 Search PubMed.
-
M. Clarembeau and P. Steylaerts, US Pat. 5
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 849974, 1998 Search PubMed.
849974, 1998 Search PubMed. - Q. Sun, M. A. Harmer and W. E. Farneth, Chem. Commun., 1996, 1201 RSC.
- A. Heidekum, M. A. Harmer and W. F. Hoelderich, J. Catal., 1999, 188, 230 CrossRef CAS.
- A. Heidekum, M. A. Harmer and W. F. Hoelderich, J. Catal., 1998, 176, 260 CrossRef CAS.
- M. A. Harmer, Q. Sun, A. Vega, W. E. Farneth, A. Heidekum and W. F. Hoelderich, Green Chem., 2000, 1, 7 RSC.
| This journal is © The Royal Society of Chemistry 2007 |