DOI:
10.1039/B604990G
(Paper)
Green Chem., 2007,
9, 49-57
FeIII–TAML-catalyzed green oxidative degradation of the azo dye Orange II by H2O2 and organic peroxides: products, toxicity, kinetics, and mechanisms†
Received
6th April 2006
, Accepted 4th October 2006
First published on 17th October 2006
Abstract
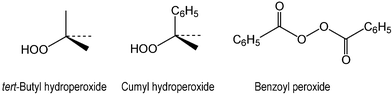
Oxidation of Orange II ([4-[(2-hydroxynaphtyl)azo]benzenesulfonic acid], sodium salt) by hydrogen peroxide catalyzed by iron(III) complexed to tetra amido macrocyclic ligands (FeIII–TAML activators) in aqueous solutions at pH 9–11 leads to CO2, CO, phthalic acid and smaller aliphatic carboxylic acids as major mineralization products. The products are non-toxic according to the Daphnia magna test. Several organic intermediates have been identified by HPLC and GC-MS that allowed the detailed description of Orange II degradation. The catalytic oxidation can also be performed by organic oxidants such as benzoyl peroxide, tert-butyl and cumyl hydroperoxides. Kinetic studies of the catalyzed oxidation indicated that FeIII–TAML activators react first with ROOR′ to form an oxidized catalyst (kI), which then oxidizes Orange II (kII). Neglecting the reversibility of the first step, the rate equation is rate = kIkII[FeIII][ROOR′][Dye]/(kI[ROOR′] + kII[Dye]); here FeIII and ROOR′ represent the catalyst and peroxide, respectively. The rate constant kI equals (74 ± 3) × 103, (1.4 ± 0.1) × 103, 24 ± 2, and 11 ± 1 M−1 s−1 for benzoyl peroxide, H2O2, t-BuOOH, and cumyl hydroperoxide at pH 9 and 25 °C, respectively. An average value of kII equals (3.1 ± 0.9) × 104 M−1 s−1 under the same conditions. The unraveling of the kinetic mechanism allows the comprehension of the robust reactivity, and this is discussed in detail using the representative results of DFT calculations.
Introduction
Effluents containing textile dyes discharged by dye manufacturing and textile wet processing industries present significant environmental problems. In 1978, 9000 tons (2%) of the 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of dyes produced worldwide was discharged in effluents from textile dyeing industries.1 The total amount of dyes discharged was about 50000 tons.1 With the worldwide growth in fiber consumption, the use of dyes and their discharge is likely to continue to rise. Modern textile dyes are chemically stable and resistant to degradation by sunlight, water, soap, bleach, and perspiration.2 They are difficult to degrade in textile wastewaters under the aerobic conditions that prevail in biological treatment plants.3,4 Dyes in wastewater create aesthetic problems, limit the possible uses of the water, and reduce the efficacy of microbiological wastewater treatment because they may be toxic to microorganisms.5 Dyes absorb and scatter the sunlight essential for the algae growth. The products of dye degradation can be mutagenic, carcinogenic, or teratogenic6 and cause long-term health concerns.7 Azo dyes that incorporate the –N
000 tons of dyes produced worldwide was discharged in effluents from textile dyeing industries.1 The total amount of dyes discharged was about 50000 tons.1 With the worldwide growth in fiber consumption, the use of dyes and their discharge is likely to continue to rise. Modern textile dyes are chemically stable and resistant to degradation by sunlight, water, soap, bleach, and perspiration.2 They are difficult to degrade in textile wastewaters under the aerobic conditions that prevail in biological treatment plants.3,4 Dyes in wastewater create aesthetic problems, limit the possible uses of the water, and reduce the efficacy of microbiological wastewater treatment because they may be toxic to microorganisms.5 Dyes absorb and scatter the sunlight essential for the algae growth. The products of dye degradation can be mutagenic, carcinogenic, or teratogenic6 and cause long-term health concerns.7 Azo dyes that incorporate the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– moiety account for up to 70% of all textile dyestuffs produced.2
N– moiety account for up to 70% of all textile dyestuffs produced.2
The degradation of organic dyes has therefore attracted much attention. New technologies for wastewater decolorization are especially needed. Modern chemical methods include TiO2-mediated photodegradation,8–11 Fenton systems,12 and soluble transition metal catalysts in combination with various oxidizing agents.13–23 Several research groups have studied the catalyzed decolorization of dyes by H2O2.24,25 Tosik and Wiktorowski have reported on the decolorization by ozone and H2O2 with Fenton reagent.26 Ozone degrades practically all dyes, reacting rapidly with both C–N and NN bonds. The reactions are slow at lower temperatures and require higher catalyst and H2O2 doses. Segal et al. have studied the oxidation of pinacyanol chloride by H2O2 in the presence of several catalysts in aqueous alkaline solution at room temperature.27 The catalysts known as octahedral molecular sieves are the most active when doped with FeIII, CrIII, and CoII. Fourteen different catalysts have been compared. The deepest bleaching (65%) was accomplished in a matter of 30 min.
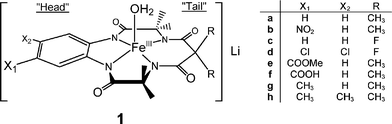 |
| | Chart 1 Iron(III)–TAML activators used in this work. | |
We have long been interested in developing green methodologies for decomposing pollutants in effluent streams and have synthesized highly active catalysts, called Fe–TAML activators (FeIII complexes of tetra amido macrocyclic ligands, 1), that exhibit the capacity to marshal hydrogen peroxide to destroy pollutants in various effluent streams.28–30 FeIII–TAML activators and their degradation products tested to date appear not to present toxicity concerns.29 Here we describe the beginnings of a detailed investigation of the use of Fe–TAML catalysts for the oxidative degradation of Orange II by H2O2 and organic peroxides. In aqueous solution, Orange II predominately exists as a keto tautomer (eqn (1)).31
| | 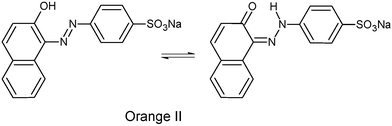 | (1) |
The reactivity of Fe–TAML activators is comparable to that of H2O2-activating enzymes.32 Therefore, it has been of interest to evaluate the relative catalytic features of FeIII–TAML activators and enzymes in oxidation of the dye. Reports on the horseradish peroxidase-catalyzed oxidation of Orange II are conflicting. According to Stiborová
et al. horseradish peroxidase does not catalyze the oxidation of Orange II by H2O2 at pH 4.7–8.4.33 Morita et al. and Zhu et al. claim that this oxidation occurs.34,35 Stoddart et al. have reported that microsomal cytochrome P450 readily catalyzes the oxidation of Orange II with a maximum activity observed at pH 7.36 Chivukula and coworkers have described the lignin peroxidase-catalyzed oxidation of Orange II into 1,2-naphthoquinone and 4-sulfophenyl hydroperoxide.37 López et al. have reported that manganese peroxidase also degrades the dye and have identified the degradation products by 1H NMR and electrospray ionization-ion trap mass spectrometry.38
Experimental
Materials
FeIII–TAML® complexes 1a–h were synthesized at Carnegie Mellon University by published methods.39 Hydrogen peroxide (30% w/w) was purchased from Fluka. Orange II, which is also called Acid Orange 7, ([4-[(2-hydroxynaphthyl)azo]benzenesulfonic acid], sodium salt) was from Aldrich. It was used without further purification (87%) for TOC measurements and studies of products of its catalytic degradation. For kinetic investigations, Orange II was purified by passing through a column filled with SMT Bulk-C18 (Separation Methods Technologies Inc., Newark) with water as the eluent. Catalase from Aspergillus niger was a Merck preparation. All other reagents and solvents (at least ACS reagent grade) were obtained from commercial sources (Aldrich, Fisher, Acros, Fluka) and used as received or, if necessary, after purification as described elsewhere.40
General procedure for degradation of Orange II by FeIII–TAML–H2O2
Orange II and 1 were combined in a buffered solution and stirred under isothermal conditions (thermostatted with a temperature precision of ±0.5 °C). Various amounts of 30% H2O2 were then added to achieve the final concentration of H2O2 of 0.01 M (Table 1). A decrease in the Orange II absorption band at the maximum (485 nm) was monitored during the reaction using a Cary 1 UV-Vis spectrophotometer. The mixture was quenched by a method appropriate to the analysis. In every experiment, the concentration of H2O2 was high enough not to constitute a limiting factor.
Analysis of degradation products by ion-chromatography
For analysis of small organic acids and ions the reaction was performed as described above and was quenched with 1.5 µL catalase (5000 U mg−1). The analysis was performed with a Dionex IonPac IC-AS50 column (250 × 2 mm) with a pre-column AG11-HC (2 × 50 mm) and a CD-25 electrochemical detector operated in a conductivity mode.
Determination of TOC
The Orange II degradation was performed as described above. The reaction was quenched with 40 µL concentrated HCl and then the solution was diluted to 5 mL with water. The sample was injected into a TOC analyzer (Schimadzu 5000) and the results for the total organic carbon in the sample (mg L−1) were obtained. Blank measurements were also performed. Blank samples consisted of 1 mL buffer, the 1a catalyst, and 40 µL HCl; the volume was then made to 5 mL with water.
Toxicity measurements
The toxicity of the dye and its degradation products were tested using 24 h born Daphnia magna at different dilutions as described in the standard operating protocol.41 Daphnids were grown in the laboratory and were fed Scenedesmus subspicatus chodat. All solutions were prepared in doubly distilled water. Room temperature was kept at 20 ± 2 °C. Toxicity experiments were carried out with 10 daphnids in each test beaker with 50 mL of final volume. Results were evaluated on the basis of immobilization percentage obtained by dividing the number of immobilized animals by total animals. The toxicity of wastewater samples was referred to as toxic when the immobilization percent was higher than 50%.41
Kinetic studies of the catalyzed bleaching of Orange II by H2O2 and organic peroxides
All kinetic measurements were performed at 298 K using a Hewlett Packard Diode Array spectrophotometer (model 8453) equipped with a thermostatted cell holder and an automatic 8-cell positioner. Stock solutions of 1 (ca. 1 × 10−3 M) and Orange II (1–8 × 10−4 M) were prepared in HPLC grade water. Solutions of hydrogen peroxide were standardized by measuring the absorbance at 230 nm (ε = 72.8 M−1 cm−1)42 or by titration as described elsewhere.43 Stock solutions of other oxidizing agents were freshly prepared daily. The pH was measured using a Corning 220 pH meter, which was calibrated with standard buffer solutions at pH 4, 7, and 10. All oxidation reactions were carried out in 0.01 M phosphate buffer at pH ranging from 5 to 11. Initial rates of Orange II oxidation were calculated from the linear absorbance versus time plots using the extinction coefficients for Orange II of 17800, 23000, and 19400 M−1 cm−1 at pH 5–7, 9 and 11, respectively, when the conversion of the dye did not exceed 10–20%. A typical kinetic run was performed as follows. The phosphate buffer was added to a cuvette with a stopper. Appropriate amounts of the stock solutions of Orange II and activator were then added. The reaction was initiated by addition of an aliquot of the stock solution of oxidizing agent. All initial rates reported are the mean values of at least three determinations. Calculations of the rate constants were carried out using a Sigma Plot 2001 package (Version 7.0).
DFT calculations
Density functional calculations were performed using Becke's three parameter hybrid functional (B3LYP) provided by the Gaussian 03 (release B.05) software package.44 The basis set 6–31G was used for geometry optimizations and Mulliken charge analysis. All calculations were carried out in vacuum (in the absence of any solvent). Mulliken population analysis was employed to monitor electron distribution. The self-consistent field calculations were terminated upon reaching tight convergence criteria (10−6 root mean square deviation in the density matrix and 10−8 atomic unit maximum deviation in energy). The calculations for benzoyl peroxide activation mechanism were performed using acetyl peroxide as a model.
Results and discussion
General observations
Electronic spectroscopy is a rational technique for monitoring the oxidation of Orange II. The dye has an intense absorption band with a maximum at 485 nm in water. Hydrogen peroxide itself oxidizes Orange II very slowly. FeIII–TAML activators increase the rate immensely; the spectral changes observed during 1a-catalyzed oxidation of Orange II are presented in Fig. 1. The major oxidation trend is a gradual decrease in the maximum intensity at 485 nm. The collapse of the 485 nm chromophore is due to the lost conjugation in the dye leading to colorless oxidation products. The inset to Fig. 1 shows that absorbance at 350 nm first increases and then decreases. This bi-phasic behavior, which is also observed for 1c but the maximum of the 350 nm band is developed faster than in the case of 1a, is obviously due to the formation and collapse of an intermediate under the catalytic conditions. The catalytic degradation performed at [H2O2] = 3.3 × 10−4 M and [1] = 2 × 10−7 M (pH 9–11, 25 °C) is usually complete within 10–30 min. Control experiments with FeIII–TAML activators in the absence of H2O2, or with H2O2 in the absence of the catalyst, or with the catalase enzyme from Aspergillus niger in the presence of H2O2, all indicated no degradation of Orange II under these experimental conditions.
![Spectral changes that accompany the catalytic oxidation of Orange II by H2O2 in the presence of 1a. Conditions: [1a] 2.6 × 10−7 M, [Orange II] 2.7 × 10−5 M, [H2O2] 4.4 × 10−4 M, pH 9 (0.01 M phosphate), 25 °C; spectra recorded with 3 min interval. Inset shows changes of absorbance at 350 nm with time; see text for details.](/image/article/2007/GC/b604990g/b604990g-f1.gif) |
| | Fig. 1 Spectral changes that accompany the catalytic oxidation of Orange II by H2O2 in the presence of 1a. Conditions: [1a] 2.6 × 10−7 M, [Orange II] 2.7 × 10−5 M, [H2O2] 4.4 × 10−4 M, pH 9 (0.01 M phosphate), 25 °C; spectra recorded with 3 min interval. Inset shows changes of absorbance at 350 nm with time; see text for details. | |
Catalysts 1a and 1c differ in terms of number of equivalents of H2O2 needed for the bleaching of one equivalent of the color of Orange II, i.e. the number of equivalents of H2O2 needed for degradation of Orange II, if its concentration is associated exclusively with the absorbance at 485 nm. Thus, a decrease of the absorbance at 485 nm has been measured after several consecutive additions of known aliquots of H2O2. Using the extinction coefficient of Orange II at 485 nm, the number of bleached equivalents has been calculated and is shown in Fig. 2. Note again that this is only the color removing stoichiometry, not the stoichiometry of complete oxidative degradation. Curiously, the 1a catalyst needs only one half equivalent of H2O2 (1 oxidation equivalent) whereas 1c needs one equivalent (2 oxidation equivalents). Hence, if only the color bleaching is at issue, the 1a catalyst is more economical because it needs half the amount of H2O2 compared to 1c. This observation may be explained if primary, non-absorbing products are oxidized slower by 1a than by 1c. Therefore, more hydrogen peroxide is used by 1c due to “spectrally invisible” oxidations (see below).
![Number of moles of Orange II bleached against number of moles of H2O2 added when catalyzed by 1a (■) and 1c (●). Conditions: [1a] 9.9 × 10−7 M, [1c] 1.24 × 10−7 M, pH 9.0 (0.01 M phosphate), 25 °C. See text for details.](/image/article/2007/GC/b604990g/b604990g-f2.gif) |
| | Fig. 2 Number of moles of Orange II bleached against number of moles of H2O2 added when catalyzed by 1a (■) and 1c (●). Conditions: [1a] 9.9 × 10−7 M, [1c] 1.24 × 10−7 M, pH 9.0 (0.01 M phosphate), 25 °C. See text for details. | |
Identification of products of degradation of Orange II
The formation of small organic acids and inorganic ions as mineralization products was anticipated. Phthalic acid was quantified using GC-MS. Formic, glycolic, and oxalic acids were detected by ion-chromatography; the data is summarized in Table 1. These breakdown molecules, known as ultimate organic products during the aromatic ring-opening,45 are non-toxic and biodegradable.46 The mineralization products are nitrite, nitrate, sulfate, and CO2 (Table 1). Nitrite and nitrate are produced from nitrogens of the azo group of Orange II. The ions NO2− and NO3− could not be reliably measured due to interference from carbonate (0.2 M). Sulfate originates from the sulfonyl group. The level of mineralization equals 35% from determining the total organic carbon.
Table 1 Mass balance (in %) after treatment of Orange II (10−4 M) by FeIII–TAML ([1a] 2 × 10−7 M), H2O2 (10−2 M) at 25 °C, pH 10 (carbonate buffer)
The data in Table 1 indicates that more than 61% of Orange II was degraded under these conditions, yielding products shown in Table 1. There may be other products, which are difficult to detect by the techniques employed. Phthalic acid, the major degradation product, is stable under the experimental conditions. It is formed from the naphthalene ring of Orange II.46,47 The presence of phthalic acid was also confirmed by GC-MS analysis (data not shown). A control test with the dye before the catalytic degradation shows no presence of this compound.
The toxicity of the solution has been evaluated before and after degradation. The data is shown in Fig. 3. The degradation products are non-toxic. The initial toxicity of Orange II decreased after the treatment by a factor of ca. 3 (see Fig. 3). Due to the protocol of the method,41 the value of mL of tested sample is a measure for toxicity. The larger this number the smaller is the toxicity. Zero points of immobilization of Daphnia magna in the control tubes were confirmed. A maximum toxicity of 20% was observed in both experiments. Zero mortality of Daphnia magna in the control tubes has been confirmed. This result demonstrates that the FeIII–TAML-catalyzed degradation of Orange II by H2O2 is safe and free from the formation of toxic oxidation products. It should be mentioned that other research groups studying peroxide catalysts employing Fenton chemistry would encounter major toxicity issues.
![Effects of Orange II (□) and its degradation products (△) on immobilization of Daphnia magna. Conditions: [Orange II] 10−4 M, [1a] 2 × 10−7 M, [H2O2] 10−2 M, 25 °C, pH 10 (0.02 M carbonate).](/image/article/2007/GC/b604990g/b604990g-f3.gif) |
| | Fig. 3 Effects of Orange II (□) and its degradation products (△) on immobilization of Daphnia magna. Conditions: [Orange II] 10−4 M, [1a] 2 × 10−7 M, [H2O2] 10−2 M, 25 °C, pH 10 (0.02 M carbonate). | |
Kinetics of 1a-catalyzed bleaching of Orange II by H2O2
The kinetic data reported in this work has been interpreted in terms of the general mechanism shown in Scheme 1. The FeIII–TAML activator reacts reversibly with a primary oxidizing agent to form an oxidized catalyst (kI, k−I), which then oxidizes a substrate (kII) followed by several fast steps.
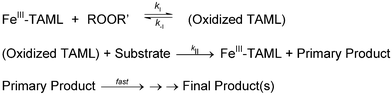 |
| | Scheme 1 General mechanism for catalysis by FeIII–TAMLs. | |
The initial rate for the consumption of Orange II is given by eqn (2), which has been derived applying the steady-state approximation to (Oxidized TAML) and using the mass balance equation for the catalyst: [FeIII]T = [FeIII] + [Oxidized TAML]. Correspondingly, the rate of bleaching should be proportional to the amount of catalyst and may level off with increasing both Orange II and oxidant concentrations. In general, these features hold for both 1a and 1c catalysts.
| | 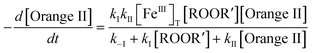 | (2) |
The initial rate method48 is the optimal kinetic technique for studying the catalytic activity of FeIII–TAML activators because 1a–h are fragile and undergo intramolecular inactivation under the operating conditions.49 This phenomenon does not compromise the catalysis but explains why, as in the case of enzymes, the kinetic curves are more difficult to analyze in their entirety, and dealing with initial rates is preferred.
![Initial rates of 1a-catalyzed bleaching of Orange II by H2O2 as a function of [1a] at pH 9 and 11. Conditions: [Orange II] 4.5 × 10−5 M, [H2O2] 3.3 × 10−4 M, 25 °C.](/image/article/2007/GC/b604990g/b604990g-f4.gif) |
| | Fig. 4 Initial rates of 1a-catalyzed bleaching of Orange II by H2O2 as a function of [1a] at pH 9 and 11. Conditions: [Orange II] 4.5 × 10−5 M, [H2O2] 3.3 × 10−4 M, 25 °C. | |
![Initial rates of 1a-catalyzed bleaching of Orange II by H2O2 as a function of [H2O2] (A) and [Orange II] (B). Conditions: 25 °C, [1a] 2 × 10−7 M, when [Orange II]fixed 4.5 × 10−4 M, [H2O2]fixed 3.3 × 10−4 M.](/image/article/2007/GC/b604990g/b604990g-f5.gif) |
| | Fig. 5 Initial rates of 1a-catalyzed bleaching of Orange II by H2O2 as a function of [H2O2] (A) and [Orange II] (B). Conditions: 25 °C, [1a] 2 × 10−7 M, when [Orange II]fixed 4.5 × 10−4 M, [H2O2]fixed 3.3 × 10−4 M. | |
The initial rate of Orange II bleaching by H2O2 is proportional to the concentration of 1a and the rate is notably higher at pH 11 than at pH 9 (Fig. 4). Minor intercepts could be attributed to the “background” non-catalytic oxidation by hydrogen peroxide. Dependencies of the initial rates on Orange II and H2O2 concentrations at pH 9 and 11 are presented in Fig. 5. It is interesting to note that at pH 11, where the bleaching is faster, there are Michaelis type dependencies in the concentrations of Orange II and H2O2. The inverse rates have linear dependence on inverse concentrations of the both reagents (graphs not shown) supporting eqn (2) and suggesting that k−I is negligible under these conditions. The case at pH 9 is slightly different. The initial rate is less curved when plotted against [H2O2] and practically independent of the Orange II concentration. To ensure that eqn (2) holds, a more detailed kinetic investigation has been performed at pH 11. The initial rates as a function of [H2O2] have been measured at different concentrations of Orange II covering at least a ten-fold concentration range. These data are shown as a three dimensional (3D) graph in Fig. 6.
![3D-Plot showing the dependence of initial rates of 1a-catalyzed bleaching of Orange II by H2O2 as a function of [H2O2] and [Orange II]. Conditions: [1a] 2 × 10−7 M, pH 11, 25 °C.](/image/article/2007/GC/b604990g/b604990g-f6.gif) |
| | Fig. 6 3D-Plot showing the dependence of initial rates of 1a-catalyzed bleaching of Orange II by H2O2 as a function of [H2O2] and [Orange II]. Conditions: [1a] 2 × 10−7 M, pH 11, 25 °C. | |
All data in Fig. 6 have been fitted to eqn (2) on the assumption that k−I
∼ 0 to obtain the rate constants kI and kII of (1.9 ± 0.2) × 103 and (4.5 ± 0.5) × 104 M−1 s−1, respectively. The 3D mesh in Fig. 6 has been generated using the best-fit rate constants; the black circles are the experimentally measured initial rates. Data in Fig. 6 agree with those in Fig. 5 and illustrate the catalytic dye bleaching by FeIII–TAML activators. When the concentration of H2O2 is low, the formation of the oxidized TAML is rate-limiting and the initial rates are independent of [Orange II]. Speeding up the formation of oxidized TAML by increasing [H2O2] moves the bleaching into a different kinetic regime, where the step driven by kII is the slowest and hence the rate is almost proportional to the Orange II concentration. Such 3D plots are observed when numerical values of the products kI[H2O2] and kII[Dye] are comparable.50
Table 2 The rate constants kI and kII (in M−1 s−1) for the 1-catalyzed oxidation of Orange II at pH 9 and 25 °C
| Catalyst (X) |
Peroxide |
10−3
×
kI |
10−4
×
kII |
|
1a (H) |
H2O2 |
1.40 ± 0.01 |
3.8 ± 0.3 |
|
1g (Me) |
H2O2 |
0.27 ± 0.04 |
1.2 ± 0.4 |
|
1h (Me2) |
H2O2 |
0.26 ± 0.02 |
1.4 ± 0.7 |
|
1f (COOH) |
H2O2 |
0.29 ± 0.01 |
1.3 ± 0.4 |
|
1e (COOMe) |
H2O2 |
0.68 ± 0.03 |
1.6 ± 0.4 |
|
1b (NO2) |
H2O2 |
1.58 ± 0.03 |
3.22 ± 0.04 |
|
1a (H) |
Benzoyl peroxide |
74 ± 3 |
3.7 ± 0.9 |
|
1a (H) |
t-BuOOH |
0.024 ± 0.002 |
3.2 ± 1.5 |
|
1a (H) |
Cumyl hydroperoxide |
0.011 ± 0.001 |
1.8 ± 0.8 |
Substituent (Hammett) effects on the rate constants kI and kII
Using a series of FeIII–TAML activators with the same R = Me but different ring substituents X, i.e. complexes 1a,b,e–h, electronic effects on the rate constants kI and kII have been investigated. The values of kI and kII have been calculated from the rates of Orange II bleaching catalyzed by 1a,b,e–h measured at H2O2 concentrations in the range (0.3–12) × 10−3 M at pH 9 and 25 °C (Table 2). Two corresponding Hammett plots are shown in Fig. 7. As seen, the electron-withdrawing groups at the aromatic ring favor both kI and kII, the effect on kI being stronger. The positive ρ values equal 0.4 and 0.2 for kI and kII, respectively. The value of 0.4 agrees well with that previously found (0.3) using an easy-to-oxidize RuII dye, for which the relation kI[H2O2] ≪
kII[Dye] holds always.32 Rather surprisingly, the sensitivity of FeIII–TAML activators to the electronic effects in the aromatic ring is exceptionally low for both kI and kII processes. The information for kII has not been reported before. The slope of 0.2 indicates no dependence. This may reflect the fact that the keto form of Orange II undergoes oxidation. If the hydroxy form would be oxidized at pH 9, much stronger electronic effects could be anticipated due to involvement of the deprotonated naphthol fragment. It is interesting to note that the data for 1a are somewhat higher than anticipated on the basis of Hammett correlations. The open circle and triangle were ignored here in the linear regressions. Similar behavior for 1a has been noted previously.49 A rather speculative explanation of this is that the less-substituted 1a catalyst has less steric restrictions when it interacts with both oxidants and electron donors.
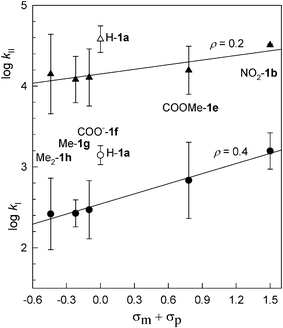 |
| | Fig. 7 Hammett plots for the rate constants kI and kII. The data for the 1a activator (Table 2) was ignored in linear regressions, the analytic expressions for which are log kI = (2.5 ± 0.02) + (0.42 ± 0.03)(σm + σp), log kII = (4.2 ± 0.04) + (0.19 ± 0.06)(σm + σp). Doubled value of (σm + σp) was used for 1h. All values of σ are taken from the review of Hansch, Leo, and Taft.57 | |
![Initial rates of 1c-catalyzed bleaching of Orange II by H2O2 at different pH. Conditions: [Orange II] 4.47 × 10−5 M, [1c] 2.5 × 10−7 M, 25 °C.](/image/article/2007/GC/b604990g/b604990g-f8.gif) |
| | Fig. 8 Initial rates of 1c-catalyzed bleaching of Orange II by H2O2 at different pH. Conditions: [Orange II] 4.47 × 10−5 M, [1c] 2.5 × 10−7 M, 25 °C. | |
Kinetics of 1c-catalyzed bleaching of Orange II by H2O2
As mentioned above, the catalysis by 1a and 1c is slightly different. The kinetic features of these activators have therefore been compared. There is a satisfactory first order kinetics in 1c in bleaching of Orange II by H2O2 at pH 5–11 (Fig. 1S in ESI†). At this point, 1a and 1c behave similarly. The rate is strongly pH-dependent and the highest activity is observed at pH 9–11. Variation in kinetic performance of 1a and 1c is revealed in Fig. 8 where the initial rate is shown as a function of [H2O2]. The rate is almost directly proportional to the peroxide concentration. The transition state is thus made of 1c and H2O2 implying that the rate should be independent of Orange II concentration. The data in Fig. 2S (see ESI†) confirms this. The activation of H2O2 by 1c is thus slower than the subsequent reaction of the oxidized TAML with Orange II, i.e.kII[Orange II] ≫
kI[H2O2]. In other words, in just bleaching, catalyst 1c is much more powerful than 1a. This also explains the different stoichiometries observed for 1a and 1c. The catalyst 1c is more robust and performs a higher number of “invisible” turnovers using extra equivalents of H2O2. Data for 1c has been used to construct pH dependence of its catalytic activity, which is actually the pH profile for the rate constant kI. The highest activities at pH 9–11 (Fig. 9) agree with our previous findings using the easiest-to-oxidize dyes and single turnover stopped-flow measurements.32,51 The behavior of 1d is similar to that of 1c.
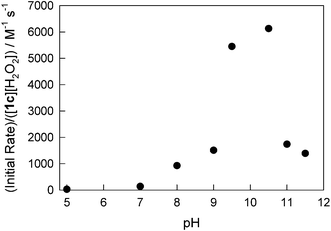 |
| | Fig. 9 Normalized initial rates of the 1c-catalyzed bleaching of Orange II measured at lower concentrations of H2O2. For conditions, see legend to Fig. 8. | |
![Initial rate of 1a-catalyzed bleaching of Orange II as a function of benzoyl peroxide concentration. Inset shows linear double reciprocal plot used for calculation of kI and kII. Conditions: [Orange II] 2.7 × 10−5 M, [1a] 1.8 × 10−8 M, pH 9, 25 °C.](/image/article/2007/GC/b604990g/b604990g-f10.gif) |
| | Fig. 10 Initial rate of 1a-catalyzed bleaching of Orange II as a function of benzoyl peroxide concentration. Inset shows linear double reciprocal plot used for calculation of kI and kII. Conditions: [Orange II] 2.7 × 10−5 M, [1a] 1.8 × 10−8 M, pH 9, 25 °C. | |
Mechanistic conclusions and implications
The mechanistic picture of FeIII–TAML-catalyzed bleaching of Orange II by a series of oxidants has two aspects. Suggested by Scheme 1 and eqn (2); these two are the intermediate formation and its performance in the dye oxidation. The steps are characterized by the rate constants kI and kII, respectively.
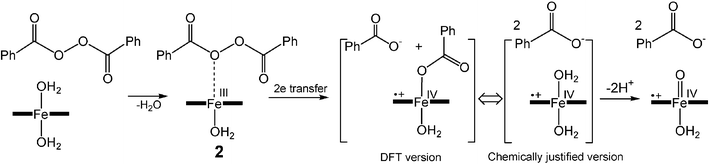
The first unexpected aspect is the highest rate constant kI for benzoyl peroxide. This molecule is significantly larger than H2O2, but the steric effect does not seem to be a factor. Assuming that the activation of hydroperoxides by FeIII–TAMLs 1a–h occurs as it has been suggested for peroxidases, i.e. involving the formation of the FeIII–O–OR intermediate followed by the heterolytic cleavage of the O–O bond,52,53 the reaction of benzoyl peroxide differs (Scheme 2). Benzoyl peroxide does not have an H–O fragment and therefore its coordination to iron(III) should involve either carbonyl or peroxo oxygen. The DFT calculations suggest that both oxygen atoms may coordinate to iron(III) because their effective negative charges are similar, i.e.
−0.38 and −0.30 for carbonyl and peroxo oxygen, respectively. The peroxo oxygen (intermediate 2 in Scheme 2) seems to be a better candidate because the σ* orbital of the O–O fragment is thus much closer to the reducing FeIII center. The DFT calculations (in vacuum) support this mechanistic hypothesis. These have been done by the example of acetyl peroxide instead of benzoyl peroxide. If the carbonyl oxygen is a donor center, the complexation between 1 and benzoyl peroxide is a dead-end pathway, which does not result in the O–O bond cleavage. In contrast, the binding to form intermediate 2 is on the reaction coordinate and the following energy minimum is found for the system consisting of free benzoate and benzoate coordinated to formally FeV (or ˙+FeIV). Benzoate coordination to iron of TAML does not appear to occur in the aqueous solution because axial ligands are known to undergo ready hydrolysis.54 Ligation of iron of TAMLs by anionic ligands is difficult to achieve in water, and therefore benzoate coordination is improbable. If formed, it will be rapidly hydrolyzed. A more realistic mechanism should involve two free benzoates and oxidized iron (Scheme 2). The formation of two benzoate anions requires two electrons. Two electrons could be moved concertedly or stepwise. The net result is the oxidation of FeIII–TAML and generation of two benzoate anions.
The major feature of the second aspect is the absolute value of the rate constants kII. The average measured value for kII in the case of 1a from Table 2 equals 3.1 × 104 M−1 s−1 at 25 °C and pH 9 (or 4.5 × 104 M−1 s−1 at pH 11). Oakes and Gratton reported the value of 0.08 M−1 s−1 for the oxidation of Orange II by p-sulfonated perbenzoic acid under the same conditions.55 The difference is a factor of ca. 4 ×105! This offers a convincing illustration of the power of FeIII–TAML catalysts.
An intriguing (and perhaps general) mechanism for the decolorization of dyes by one-electron oxidizing agents has been proposed after studying Orange II bleaching by pulse radiolysis.56 It involves diffusion-controlled disproportionation of two primary dye-radicals formed after the initial, rate-limiting electron transfer. The diffusion-controlled disproportionation affords a starting dye and the product of its 2-electron oxidation, which is further involved in a series of transformations. Early steps of this mechanism are shown in Scheme 3.56 This mechanism is useful for describing the results of catalysis by 1 assuming that the oxidized catalysts 1 generate the primary radical 3 shown in Scheme 3 in a pathway described by the rate constant kII.
We did not detect 4-diazenylbenzosulfonate (4) and 1,2-naphthoquinone by HPLC under our catalytic conditions. The probable reason that the quinone was not detected is that under these conditions 1,2-naphthoquinone is rapidly oxidized into phthalic acid plus two additional products just by H2O2 without participation of 1. 4-Diazenylbenzosulfonate should be unstable as well. A formation of 4-nitrobenzo- and 4-hydroxybenzosulfonate detected by HPLC could presumably result from the oxidation of 4-diazenylbenzosulfonate. Phthalic acid and 4-hydroxybenzosulfonate undergo the catalyzed oxidation by H2O2 giving a variety of small biodegradable, non-toxic organic products with substantial mineralization.
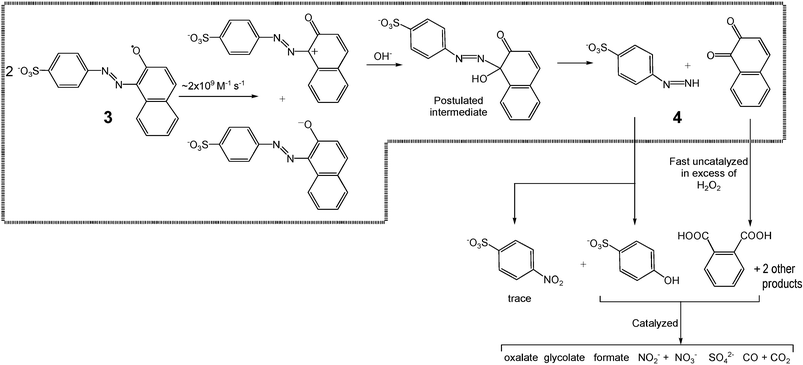 |
| | Scheme 3 Plausible scheme of 1-catalyzed oxidative degradation of Orange II. Steps in the dotted box were suggested previously.56 | |
In conclusion, the FeIII–TAML–H2O2 system efficiently degrades Orange II into small biodegradable and non-toxic organic products. Substantial mineralization has been observed. The oxidation occurs rapidly compared to biological and other chemical systems that have been previously studied. Furthermore, the ecotoxicological examination suggests that the described technique is an environmentally safe process. With current concerns regarding the treatment of textile dyes, a green process for degrading pollutants is greatly needed; this is especially true for textile dyes. The Fe–TAML–H2O2 systems show considerable promise to provide such a technology.
Acknowledgements
Support from Eden-Hall Foundation, NSF (9612990), EPA (Grant RD83), and the Institute for Green Oxidation Chemistry is gratefully acknowledged.
References
-
Pollution Prevention and Abatement Handbook, World Bank Group, Washington, D.C., 1998, ISBN 0-8213-3638-X Search PubMed.
-
H. Zollinger, Color Chemistry: Syntheses, Properties and Applications of Organic Dyes and Pigments, Wiley, Weinheim, 3rd edn, 2003 Search PubMed.
-
J. R. Easton, in The dye maker's view of color in dye house effluent, ed. P. Cooper, Woodhead Publishing Ltd, Bradford, UK, 1995 Search PubMed.
- A. Gottlieb, C. Shaw, A. Smith, A. Wheatley and S. Forsythe, J. Biotechnol., 2003, 101, 49 CrossRef CAS.
- K. Hunger, Chimia, 1994, 48, 520; K. Hunger, Rev. Prog. Color. Relat. Top., 2005, 35, 76 Search PubMed.
- C. Jakopitsch, G. Regelsberger, P. G. Furtmueller, F. Rueker, G. A. Peschek and C. Obinger, Biochem. Biophys. Res. Commun., 2001, 287, 682 CrossRef CAS.
- M. A. Brown and S. C. DeVito, Crit. Rev. Environ. Sci. Technol., 1993, 23, 249 Search PubMed.
- C. Lagrasta, I. R. Bellobono and M. Bonardi, J. Photochem. Photobiol., A, 1997, 110, 201 CrossRef CAS.
- C. A. K. Gouvea, F. Wypych, S. G. Moraes, N. Duran, N. Nagata and P. Peralta-Zamora, Chemosphere, 1999, 40, 433.
- B. Neppolian, S. Sakthivel, B. Arabindoo, M. Palanichamy and V. Murugesan, J. Environ. Sci. Health, Part A, 1999, A34, 1829 Search PubMed.
- B. Neppolian, H. C. Choi, S. Sakthivel, B. Arabindoo and V. Murugesan, Chemosphere, 2002, 46, 1173 CrossRef CAS.
- B. Utset, J. Garcia, J. Casado, X. Domenech and J. Peral, Chemosphere, 2000, 41, 1187 CrossRef CAS.
- J. Oakes, P. Gratton and I. Weil, J. Chem. Soc., Dalton Trans., 1997, 3805 RSC.
- J. Oakes, G. Welch and P. Gratton, J. Chem. Soc., Dalton Trans., 1997, 3811 RSC.
- J. Oakes and P. Gratton, J. Chem. Soc., Perkin Trans. 2, 1998, 2563 RSC.
- J. Oakes, P. Gratton, R. Clark and I. Wilkes, J. Chem. Soc., Perkin Trans. 2, 1998, 2569 RSC.
- G. R. Hodges, J. R. Lindsay Smith and J. Oakes, J. Chem. Soc., Perkin Trans. 2, 1998, 617 RSC.
- G. R. Hodges, J. R. Lindsay Smith and J. Oakes, J. Chem. Soc., Perkin Trans. 2, 1999, 1943 RSC.
- I. Arslan and I. A. Balcioglu, Dyes Pigm., 1999, 43, 95 CrossRef CAS.
- J. Oakes, P. Gratton and T. Gordon-Smith, Dyes Pigm., 2000, 46, 169 CrossRef CAS.
- B. C. Gilbert, J. R. L. Smith, M. S. Newton, J. Oakes and R. Pons i Prats, Org. Biomol. Chem., 2003, 1, 1568 RSC.
-
R. A. Sheldon and G. Papadogianakis, in Aqueous-Phase Organometallic Catalysis, ed. B. Cornils and W. A. Herrmann, Wiley-VCH Verlag, Weinheim, 2nd edn, 2004, pp. 473–480 Search PubMed.
- R. Hage and A. Lienke, Angew. Chem., Int. Ed., 2006, 45, 206 CrossRef CAS.
- D. M. Gould, W. P. Griffith and M. Spiro, J. Mol. Catal. A: Chem., 2001, 175, 289 CrossRef CAS.
- P. Verma, P. Baldrian and F. Nerud, Chemosphere, 2003, 50, 975 CrossRef CAS.
- R. Tosik and S. Wiktorowski, Ozone: Sci. Eng., 2001, 23, 295 Search PubMed.
- S. R. Segal, S. L. Suib and L. Foland, Chem. Mater., 1997, 9, 2526 CrossRef CAS.
-
T. J. Collins and C. P. Horwitz, TAPPI Pulping Conference, Orlando, Fl, Oct. 31–Nov. 4, 1999, p. 703 Search PubMed.
- S. Sen Gupta, M. Stadler, C. A. Noser, A. Ghosh, B. Steinhoff, D. Lenoir, C. P. Horwitz, K.-W. Schramm and T. J. Collins, Science, 2002, 296, 326 CrossRef.
- T. J. Collins, Acc. Chem. Res., 2002, 35, 782 CrossRef CAS.
- L. C. Abbott, S. N. Batchelor, J. Oakes, B. C. Gilbert, A. C. Whitwood, J. R. L. Smith and J. N. Moore, J. Phys. Chem., 2005, 109, 2894 Search PubMed.
-
A. Ghosh, D. A. Mitchell, A. D. Ryabov, D. L. Popescu, A. Chanda and T. J. Collins, manuscript in preparation.
- M. Stiborova, B. Asfaw, E. Frei and H. H. Schmeiser, Collect. Czech. Chem. Commun., 1996, 61, 962 CrossRef CAS.
- M. Morita, E. Yamaguchi, E. Komatsu, R. Ito, T. Kamidate and H. Watanabe, Nihon Yukagakkaishi, 1999, 48, 793 Search PubMed.
- M. Zhu, X.-M. Huang and H.-X. Shen, Chin. J. Chem., 1999, 17, 356 CAS.
- A. M. Stoddart and W. G. Levine, Biochem. Pharmacol., 1992, 43, 2227 CrossRef CAS.
- M. Chivukula, J. T. Spadaro and V. Renganathan, Biochemistry, 1995, 34, 7765 CrossRef CAS.
- C. López, A.-G. Valade, B. Combourieu, I. Mielgo, B. Bouchon and J. M. Lema, Anal. Biochem., 2004, 335, 135 CrossRef CAS.
- Website: http://www.chem.cmu.edu/groups/collins/awardpatpub/patents/index.html.
-
D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon Press, 1988 Search PubMed.
-
DIN EN ISO/IEC 17025, General Requirements of the Authority of Test and Calibration Laboratories. Version in German, English, French, CEN/CENELEC, Brussels, Belgium, 2000; Normausschuss Wasserwesen (NAW) beim Deutschen Institut für Normung e.V. (DIN). Arbeitsgruppe Biotest: Testverfahren mit Wasserorganismen (Gruppe L): (a) Bestimmung der Wirkung von Wasserinhaltsstoffen auf Kleinkrebse, Prüfungsvorschrift DIN 38 412 Teil 11; (b)Allgemeine Hinweise zur Planung, Durchführung und Auswertung biologischer Testverfahren, Prüfvorschrift DIN 38 412 Teil 1. OECD 202 Guideline for Testing of Chemicals 202. Daphnia sp., Acute Immobilisation Test and Reproduction Test, OECD, Paris 1984 Search PubMed.
- P. George, Biochem. J., 1953, 54, 267 CAS.
- V. N. Goral, M. I. Nelen and A. D. Ryabov, Anal. Lett., 1995, 28, 2139 CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision B.05), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- J. Bandara and J. Kiwi, New J. Chem., 1999, 23, 717 RSC.
- M. F. Coughlin, B. K. Kinkle and P. L. Bishop, Chemosphere, 2002, 46, 11 CrossRef CAS.
- V. Nadtochenko and J. Kiwi, J. Chem. Soc., Faraday Trans., 1997, 93, 2373 RSC.
-
J. H. Espenson, Chemical Kinetics and Reaction Mechanisms, McGraw-Hill, Inc., New York, 1995.
- A. Chanda, A. D. Ryabov, S. Mondal, L. Alexandrova, A. Ghosh, Y. Hangun-Balkir, C. P. Horwitz and T. J. Collins, Chem.–Eur. J., 2006 DOI:10.1002/chem.200600630.
- A. D. Ryabov, V. S. Sukharev, L. Alexandrova, R. Le Lagadec and M. Pfeffer, Inorg. Chem., 2001, 40, 6529 CrossRef CAS.
-
D.-L. Popescu, A. D. Ryabov and T. J. Collins, Abstracts of Papers, 231st ACS National Meeting, Atlanta, GA, US, March 26–30, 2006, p. IEC-052.
- H. B. Dunford, Adv. Inorg. Biochem., 1982, 4, 41 CAS.
-
H. B. Dunford, Heme Peroxidases, Wiley-VCH, New York, 1999 Search PubMed.
- A. Ghosh, A. D. Ryabov, S. M. Mayer, D. C. Horner, D. E. Prasuhn, Jr., S. Sen Gupta, L. Vuocolo, C. Culver, M. P. Hendrich, C. E. F. Rickard, R. E. Norman, C. P. Horwitz and T. J. Collins, J. Am. Chem. Soc., 2003, 125, 12378 CrossRef CAS.
- J. Oakes and P. Gratton, J. Chem. Soc., Perkin Trans. 2, 1998, 1857 RSC.
- J. J. F. Coen, A. T. Smith, L. P. Candeias and J. Oakes, J. Chem. Soc., Perkin Trans. 2, 2001, 2125 RSC.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2007 |
Click here to see how this site uses Cookies. View our privacy policy here.