Photo-driven molecular devices
Sourav
Saha
and
J. Fraser
Stoddart
*
Department of Chemistry and Biochemistry and California NanoSystems Institute, University of California, Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095. E-mail: stoddart@chem.ucla.edu; Fax: (+1) 310-206-5621; Tel: (+1) 310-206-7078
First published on 15th November 2006
Abstract
In this critical review, we discuss switching of the light-powered bistable rotaxanes and catenanes and highlight the practical applications of some of these systems. Photoactive molecular and supramolecular machines are comprised of two parts—1) a switching element, based on noncovalent interactions within the recognition units, which is responsible for executing mechanical movement, and 2) a light-harvesting unit which utilizes light to control the competitive interactions between the recognition sites. We also survey another class of molecular devices, namely molecular rotary motors—i.e., those that behave like their macroscopic counterparts—in which photochemically and thermally induced mechanical movement relies on isomerizations of a pivotal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, leading to a rotation of the top propeller part with respect to the stationary bottom part of the helical shaped chiral molecule. (146 references.)
C bond, leading to a rotation of the top propeller part with respect to the stationary bottom part of the helical shaped chiral molecule. (146 references.)
 Sourav Saha | Sourav Saha received an MSc degree in Chemistry from the Indian Institute of Technology in Kanpur in 2000 and a PhD degree from the University of California, Los Angeles (UCLA) in 2005 working on Light and Electron Powered Molecular and Supramolecular Machines. Presently, he is a postdoctoral researcher in Professor Fraser Stoddart's Supramolecular Chemistry Laboratory at UCLA. He is working towards developing bistable rotaxane-based molecular logic-gates in crossbar electrode architectures as well as new generations of photo-driven molecular shuttles as prototypes of molecular devices and machines. |
 J. Fraser Stoddart | J. Fraser Stoddart was born in Edinburgh, Scotland, in 1942. He received all (BSc, PhD, and DSc) of his degrees from the University of Edinburgh, U.K. Presently, he holds the Fred Kavli Chair in NanoSystems Sciences at UCLA and is the Director of the California NanoSystems Institute. His current research interests are concerned with transporting well-established concepts in biology from the life sciences into materials and medicinal chemistry. |
1. Introduction
Machines need power supplies for them to undergo mechanical movements and hence do work. Just as macroscopic machines require macroscopic energy sources, natural1–6 and artificial molecular machinery7–37 require nanoscale power supplies to undergo directed molecular motions. While macroscopic machines do not move until energy is supplied to them for a specific function, nanoscale machines down at the molecular level are in perpetual Brownian motion at ambient temperatures. Thus, for machine-like functions to be expressed by molecular systems, they should be able to execute specific and directional mechanical movements—when stimulated by external inputs—that are otherwise only of a haphazard nature on account of Brownian motion . The intrinsic random motions of molecular machines make the detection of the stimuli-induced, relative movements of their components a nontrivial phenomenon, as compared to the easily detectable, obvious motions executed by macroscopic machines. Three types of external stimuli—that is chemical,38–45 electrochemical,46–54 and photochemical54–64—have been employed to induce well-defined mechanical movements within molecular machines. Several spectroscopic and electrochemical techniques have been introduced to distinguish the stimuli-induced nanomechanical motions from the random Brownian ones.Mechanically interlocked molecules, such as bistable [2]catenanes65–67 and [2]rotaxanes68–76 are some of the most suitable candidates to serve as nanoscale mechanical switches in the rapidly evolving field of nanoelectronics77–88 and nanoelectromechanical systems89–100 (NEMS). The advantages of mechanically interlocked molecules for potential applications in the realm of molecular electronics stem from their abilities to undergo101–128 controllable nanoelectromechanical motions. In bistable catenanes and rotaxanes , changing the affinity of the ring for the two distinct recognition sites on the dumbbell component allows the ring to be translated between the sites and thus relative mechanical movement is produced. A minimum condition for generating such a translational motion within a bistable [2]rotaxane is a large difference in the binding affinities for the ring by the two different recognition sites on the dumbbell component in the ground state . Thereafter, it is important that these relative binding affinities can be reversed by an external stimulus. A free energy difference of greater than 1.2 kcal mol–1 between the two translational isomers ensures51 that, at room temperature, 90% of the ring is positioned around one recognition site in preference to the other one, a situation which gives rise to a highly preferred ground state co-conformation (GSCC). Modification of the binding affinity of the primary recognition site by an external stimulus38–64 turns OFF the stabilizing interactions in the GSCC and triggers the translational motion of the ring component to the alternative recognition site which provides temporarily better stabilizing interactions. The ring's location around the second recognition site, even after the removal of the external stimulus and the re-instatement of the GSCC, results in an isoelectronic translational isomer of the bistable rotaxane , which is defined as the metastable state co-conformation (MSCC). Relaxation of the MSCC to reproduce the GSCC, triggered by either the ambient thermal energy or another stimulus, completes the full cycle of the machine-like movement of bistable rotaxanes , making them one of the most accessible molecular machines.
2. Light-powered linear motion within a metal ion-based rotaxane
The transition metal based [2]rotaxane 1+/2+ is composed129 (Fig. 1) of a 30-membered ring (M30) containing a bidentate 2,9-diphenyl-1,10-phenanthroline (phen) ligand, which is interlocked onto a dumbbell-shaped component, equipped with another 2,9-disubstituted phen ligand in addition to a terdentate 2,2′,6′,2′′-terpyridine (terpy) ligand. The ligands on the ring and dumbbell components chelate differently with CuI and CuII ions, depending on the oxidation state of the cation. The ring can be switched between the phen and terpy ligands on the dumbbell component by controlling the oxidation states of the complexed Cu ions and hence, their coordination environments. The CuI(4) state favors a tetrahedral geometry, a requirement which is satisfied when the bidentate phen ligands from both the ring and the dumbbell are involved in chelation. By contrast, the pentacoordination of the CuII(5) state is satisfied by the involvement of the phen ligand from the ring and the terpy ligand from the dumbbell component. Photochemical or electrochemical oxidation of the complexed CuI(4) to CuII(5) drives the ring from the bidentate phen site to the terdentate terpy site along the dumbbell component and vice versa. Irradiation of the [2]rotaxane 1+-CuI with 464 nm light in the presence of an electron acceptor (p-nitrobenzyl bromide) generates 12+-CuII(4), which relaxes to the stable 12+-CuII(5) on account of the mechanical movement of the ring to the terpy site. Addition of a reductant (ascorbic acid) reduces the CuII(5) to the CuI(4) state, which, in turn, triggers the return of the ring to the phen site on the dumbbell. As a result, the movement of the ring can be powered by hybrid photochemical–chemical stimuli. The light-driven switching of the rotaxane 1+/2+ was found to be completely reversible in the presence of tetrabutylammonium tetrafluoroborate (TBABF4). A direct electrochemical redox process involving the CuI(4)/CuII(5) couple also induces a similar translational motion in the bistable [2]rotaxane.![Photochemically induced switching of a metal complex-based bistable [2]rotaxane 1+/12+. Photo-induced oxidation of the Cu(i) center to Cu(ii) drives the bidentate macrocycle M30 to the terdentate terpy unit of the dumbbell component, whereas a chemical reduction of Cu(ii) to Cu(i) with ascorbic acid returns macrocycle M30 to the original bidentate phen unit, completing a cyclic process which relies on changes in the Cu(i/ii) coordination geometry. (Redrawn with permission from ref. 129. Copyright (1999) American Chemical Society.)](/image/article/2007/CS/b607187b/b607187b-f1.gif) | ||
| Fig. 1 Photochemically induced switching of a metal complex-based bistable [2]rotaxane 1+/12+. Photo-induced oxidation of the Cu(I) center to Cu(II) drives the bidentate macrocycle M30 to the terdentate terpy unit of the dumbbell component, whereas a chemical reduction of Cu(II) to Cu(I) with ascorbic acid returns macrocycle M30 to the original bidentate phen unit, completing a cyclic process which relies on changes in the Cu(I/II) coordination geometry. (Redrawn with permission from ref. 129. Copyright (1999) American Chemical Society.) | ||
3. Light-driven rotational motion of a metal ion-based catenane
Circumrotary motion in a photoactive Ru(II)-complexing [2]catenane 22+ (Fig. 2), consisting130 of a 42-membered ring (M42) which contains a 2,2′-bipyridine (bipy) ligand, interlocked with a 63-membered ring (M63) containing two 1,10-phenanthroline (phen) ligands, can be powered by light and heat. In the ground state of the catenane 22+, the interlocked M42 and M63 rings are oriented in such a way that all three bidentate ligands in the two rings participate in complexation with the Ru(II) ion to satisfy its octahedral coordination environment, producing the characteristic red color of the Ru(II) complex. Upon irradiation of 22+ with a 250 W halogen lamp (λ > 300 nm) either in MeCN or in CH2Cl2 in the presence of tetraethylammonium chloride, the color of the solution turns from red (λmax = 458 nm) to purple (λmax = 561 nm) with a clean isosbestic point at 484 nm. This spectroscopic change, together with the 1H NMR chemical shift130 of the diagnostic peaks, suggests the photochemical decoordination of the M42 ring from the Ru(II) center, while two Cl− ions or MeCN molecules complex with the Ru(II) ion to generate 2′a or 2′b2+, respectively. Heating of the solution of 2′a or 2′b2+ transforms it back into the original isomer 22+. Based on the absorption and 1H NMR spectra, the photochemical decoordination and thermal recomplexation occur with >95% efficiency. Thus, the bistable catenane 22+ represents a prototype light/heat-driven artificial molecular machine, which performs rotary motion.![Photochemically induced switching of a metal complex-based bistable [2]catenane 22+. Irradiation of the catenane 22+ at λ ≥ 300 nm causes MLCT to the phen ligand of the macrocycle M42 and induces its circumrotation along the larger macrocycle M63 in the presence of (a) Cl– or (b) MeCN ligands which now occupy the empty coordination sites of the Ru(ii) ion to produce 2′a or 2′b2+, respectively. Heating of these systems regenerates the original state 22+. (Redrawn with permission from ref. 130. Copyright (2004) Wiley-VCH.)](/image/article/2007/CS/b607187b/b607187b-f2.gif) | ||
| Fig. 2 Photochemically induced switching of a metal complex-based bistable [2]catenane 22+. Irradiation of the catenane 22+ at λ ≥ 300 nm causes MLCT to the phen ligand of the macrocycle M42 and induces its circumrotation along the larger macrocycle M63 in the presence of (a) Cl– or (b) MeCN ligands which now occupy the empty coordination sites of the Ru(II) ion to produce 2′a or 2′b2+, respectively. Heating of these systems regenerates the original state 22+. (Redrawn with permission from ref. 130. Copyright (2004) Wiley-VCH.) | ||
4. Photo-driven shuttling in two constitutionally isomeric bistable rotaxanes
A multicomponent [2]rotaxane 36+ has been designed (Fig. 3a) to work56 as a visible light-driven molecular abacus. The bistable rotaxane of the Mark I generation is composed of six molecular modules, namely (i) a bis-para-phenylene[34]crown-10 (BPP34C10) electron-rich macrocycle M and a dumbbell-shaped component which contains (ii) a Ru(II)-bipyridine based light-harvesting unit P2+ as one of its stoppers, (iii) a p-terphenyl-type ring system as a rigid spacer S which separates the P2+ unit from the mechanical switching moiety comprising (iv) a 4,4′-bipyridinium (A12+) as a strong primary π-electron accepting unit and (v) 3,3′-dimethyl-4,4′-bipyridinium (A22+) as a weak secondary π-electron accepting unit that can play the role of stations for the macrocycle M, and (vi) a tetraarylmethane group T as the second stopper. In the Mark Irotaxane 36+, the A1 station is situated farthest away from the P2+ unit and is separated by the A22+ station. In the GSCC, the M encircles the stronger π-accepting A12+ station. The BPP34C10 macrocycle can be switched mechanically between the A12+ and A12+ stations with light in the presence of 1) an external reductant/oxidant—sacrificial mechanism54 (Fig. 3b), and 2) a dual-action redox reagent—autonomous mechanism61 (Fig. 3c).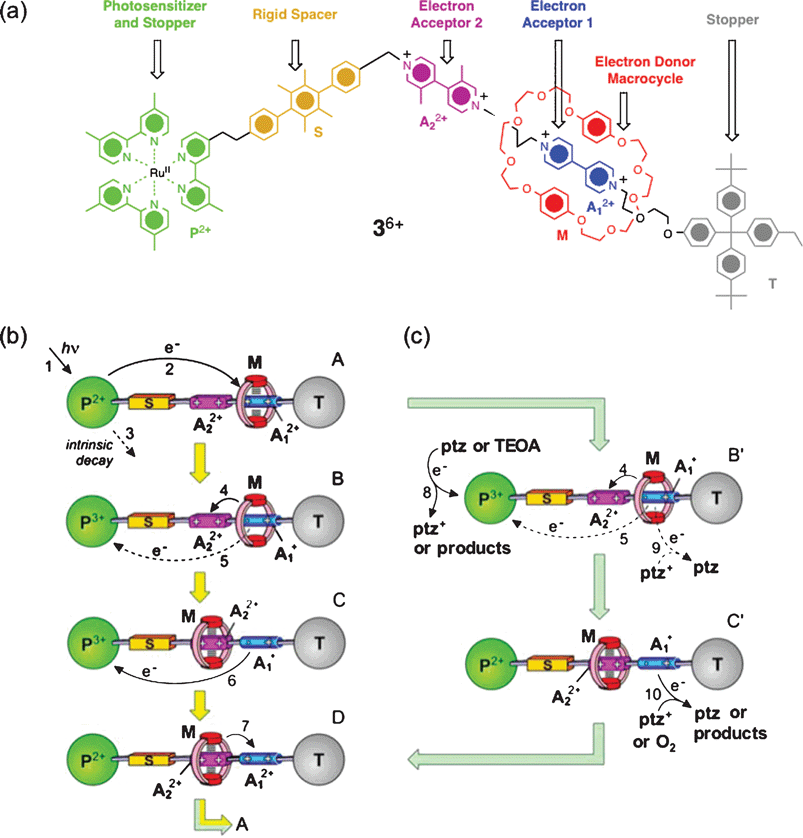 | ||
| Fig. 3 (a) Structural formula of a photoactive molecular abacus 36+. (b) Photo-driven switching process of 36+ in the presence of triethanolamine (TEOA) as an external fuel—a sacrificial mechanism: 1) photosensitization of the Ru(II) complex, 2) PET , 3) intrinsic decay, 4) forward movement of M, 5) BET —does not occur in the presence of TEOA, 6) charge recombination , 7) return of M to the A12+ unit. (c) Photo-driven switching process of 36+ in the presence of phenothiazine (ptz) as a donor–acceptor couple—an autonomous mechanism: 8) reduction of the photogenerated P3+ unit by ptz, 4) forward movement of BPP34C10, 5) no BET in the presence of ptz, 9) and 10) oxidation of the A1+radical cation back to A12+ by ptz+. (Redrawn with permission from ref. 63. Copyright (2006) National Academy of Sciences USA.) | ||
Sacrificial mechanism: Photosensitization (step 1) of the Ru(bipy)2+ complex with 532 nm light generates56 its metal-to-ligand charge-transfer (MLCT) excited state *P2+, which undergoes a photo-induced electron transfer (PET , step 2) to the stronger π-electron accepting A12+ unit, reducing it to a A1+radical cation (Fig. 3b). In the presence of an external electron donor (D), triethanolamine (TEOA), which reduces the photo-oxidized P3+ unit to prevent the back electron transfer (BET ) process (step 5), the A1+radical cation enjoys enough of a lifetime to repel (step 4) the π-electron rich macrocycle M to the weaker π-electron accepting A22+ unit. Oxidation of the A1+radical cation back to the A12+ unit by dioxygen generates the MSCC (Fig. 3b, D), which is represented by the BPP34C10 macrocycle's location around the A22+ unit. Finally, thermally assisted Brownian motions allow M to return to its original A12+ unit (step 7), completing the mechanical switching cycle in the presence of the sacrificial donor TEOA, which degrades into by-products. Thus, in order to cycle the photochemical switching process of 36+ repetitively, a stoichiometric amount of TEOA is essential for every single cycle.
Autonomous mechanism: The very same series of photochemically induced mechanical switching in 36+ was achieved63 (Fig. 3c) using phenothiazine (PTZ) as a dual-action redox reagent, which directly reduces the P3+ unit, allowing M enough time to move to the A22+ station when A12+ is photo-reduced to A1+. The reduction potential of PTZ+/0 matches very well with the oxidation potential of the A1+/2+ station, allowing a dark-phase electronic recombination to regenerate A12+. At this point, the M returns to the A12+ station, completing an autonomous switching cycle, a process in which no additional redox reagent is needed63 for the subsequent photochemical cycles.
A photoactive molecular abacus 46+ of the Mark II generation64 (Fig. 4) has been synthesized which retains all six modules used in 36+. The locations, however, of the A12+ and A22+ electron acceptors on its dumbbell component have been swapped.64 In this isomer, the macrocycle M still encircles the stronger π-electron acceptor A12+ in the GSCC which is now located closer to the light-harvesting P2+ moiety than it was in the Mark I version. Closer proximity between the P2+ and A12+ units not only accelerates the photo-induced electron transfer (PET ) process from the *P2+ unit to the A12+ unit, but also has a similar effect on the back electron transfer (BET ) process between the photogenerated P3+ and the A1+radical cation . Therefore, the lifetime of the A1+radical cation is much shorter62 in 46+ than in 36+, a feature that prevents M from moving back to the A22+ station. This issue, however, could be circumvented64 by employing TEOA as a sacrificial electron donor to reduce P3+ before BET takes place, and thus, allowing M to move to the A22+ station. Upon oxidation of the A1+radical cation back to the A12+ state by dioxygen, M returns to the A12+ by thermally assisted Brownian motion s to complete the cycle.
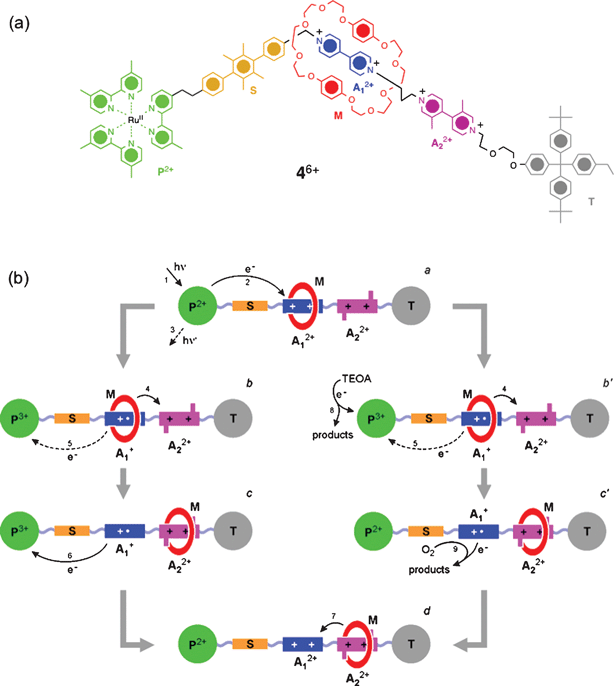 | ||
| Fig. 4 (a) Structural formula of a photoactive molecular abacus 46+ which is a constitutional isomer of the rotaxane 36+. (b) Photo-driven switching process of 46+ in the presence of TEOA—sacrificial mechanism: 1) photosensitization of the Ru(II) complex, 2) PET , 3) intrinsic decay, 4) forward movement of M, 5) BET —does not occur in the presence of TEOA, 6) charge recombination , 7) return of M to the A12+ unit, 8) reduction of the photogenerated P3+ unit by TEOA, 9) oxidation of the A1+radical cation back to A12+ by O2. (Redrawn with permission from ref. 64. Copyright (2006) CSIRO Publishing, http://www.publish.csiro.au/journals/ajc.) | ||
Since the sacrificial donor prevents BET in both Mark II and I versions of these bistable rotaxanes , it has been observed63,64 that the fuel-assisted photo-driven shuttling is more efficient in the Mark Irotaxane than in the Mark II generation on account of the faster PET in the former. The only other difference in the fuel-assisted, light-driven mechanical shuttling in these two rotaxanes is the opposite directions of the macrocycles' movements—while in the Mark Irotaxane , M moves towards the P2+ unit, it moves away from the P2+ in the Mark IIrotaxane . In both cases, however, M moves to the secondary electron accepting A22+ station, when A12+ is photo-reduced to the A1+radical cation .
5. A Lockable light-driven molecular shuttle
One of the simplest examples of photoactive molecular switches relies on light-activated cis–trans isomerization to effect switching. The bistable [2]rotaxane (E)-5 (Fig. 5) composed131 of an α-cyclodextrin (α-CD) ring, which is interlocked onto a dumbbell-shaped component containing two recognition sites—a stronger binding stilbene (S) unit and a weaker biphenyl (P) one—and terminated with two different stoppers—a 4-amino-3,6-disulfonic-1,8-naphthalimide disodium salt (N)-based stopper attached to the P unit and an isophthalic acid (I) stopper attached to the stilbene unit, has been synthesized via a Pd-catalyzed Suzuki coupling to produce the biphenyl unit in the presence of α-CD. In the ground state when the stilbene unit is predominantly in the (E)-configuration, the α-CD ring encircles the S unit—a translational isomer which is also stabilized by intramolecular hydrogen-bonding between the carboxylic acid groups on the stopper I and the secondary alcohol groups on the α-CD—preventing the light-induced E–Zphotoisomerization of the S unit. In the presence of an excess of base (Na2CO3), the isophthalic acid is deprotonated to produce (E)-52–, a process that unlocks the hydrogen-bonding interactions and allows the α-CD to move to the P unit upon photoisomerization of the S unit to the (Z)-isomer at 335 nm. The movement, as well as precise location of the α-CD driven by E–Zphotoisomerization of the stilbene unit, were investigated by 1H NMR, absorption, and emission spectroscopies, allowing a clean detection of the light-driven mechanical process. Z→E Photoisomerization at 280 nm brings the α-CD back to the S unit, completing the cycle. In the (E)-configuration of stilbene unit, the rotaxane 52– shows a weak fluorescence band at 530 nm. The emission intensity, however, rises in the (Z)-isomer when the α-CD moves back to the P unit, a process which can be activated instantaneously by simply switching the excitation wavelengths between 335 and 280 nm.![Photo-induced mechanical switching of a α-CD-based lockable [2]rotaxane (E/Z)-5 in the presence of Na2CO3.](/image/article/2007/CS/b607187b/b607187b-f5.gif) | ||
| Fig. 5 Photo-induced mechanical switching of a α-CD-based lockable [2]rotaxane (E/Z)-5 in the presence of Na2CO3. | ||
6. Photochemically and thermally driven azobenzene–α-CD rotaxane
The bistable [2]rotaxane (E/Z)-64+ (Fig. 6)—composed132 of an α-CD ring interlocked onto a dumbbell component consisting a photoisomerizable azobenzene unit as a primary station with two bismethylene units playing the roles of secondary stations for the α-CD and two bipyridinium units acting as polar stoppers for the hydrophobic cavity of the α-CD with two 2,4-dinitrophenyl units as steric stoppers for the α-CD ring—constitutes a light and thermally driven molecular shuttle. In the ground state , the α-CD ring encircles the trans-configurated azobenzene unit, which can be photoisomerized to the cis-configuration by UV-irradiation (360 nm), at which point α-CD moves mechanically away from it to one of the adjacent bismethylene stations. A cis- to trans- photoisomerization with visible light (430 nm) or by thermal isomerization of the azobenzene station brings the α-CD back to its original position. The photo-driven mechanical movement of (E/Z)-64+ in H2O and Me2SO has been investigated132 by 1H NMR and absorption spectroscopies as well as induced circular dichroism (ICD).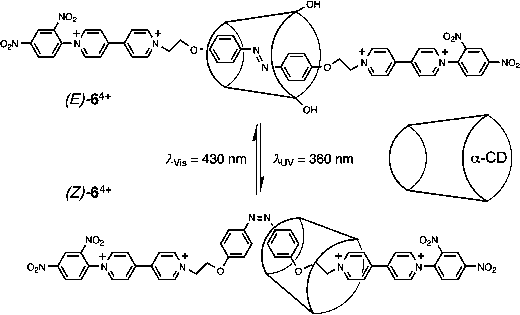 | ||
| Fig. 6 Mechanical switching of α-CD ring by photo-isomerization of the encircled (E/Z)-azobenzene-based recognition unit in the rotaxane 64+. | ||
7. Mechanical switching induced by photoisomerization
The [2]rotaxane (E/Z)-7-H22+ (Fig. 7) incorporates133 a fumaramide–maleamide unit—a photoisomerizable alkene diamide that also plays the role of a strong hydrogen-bond-acceptor in its (E)-form (fumaramide) to the surrounding hydrogen-bond donating tetramide macrocycle in (E)-7-H22+. A second hydrogen-bond accepting glycylglycine (Gly-Gly) unit and a nearby anthracene unit to act as a fluorescent label are also present in the dumbbell component of the rotaxane . The tetramide-based macrocycle provides four hydrogen-bond donating centers. Two pyridinium units in the macrocycle are known to quench the anthracene-based fluorescence through electron transfer when they are in close proximity. Irradiation of the (E)-7-H22+ isomer with 312 nm light converts the (E)-fumaramide into its (Z)-isomer, maleamide which has a significantly lower hydrogen-bond accepting ability. This change in the binding affinity allows the macrocycle to move to the intermediate hydrogen-bond accepting station Gly-Gly, generating the (Z)-7-H22+ translational isomer. Heating of the (Z)-7-H22+ isomer at 115 °C regenerates the fumaramide unit and the macrocycle returns to its original binding site in (E)-7-H22+. The longer distance between the anthracene fluorescent tag and the macrocycle containing the two pyridinium units in the (E)-7-H22+ isomer allows this isomer to fluoresce (λexc = 365 nm), whereas the anthracene fluorescence in the (Z)-7-H22+rotaxane is quenched by close proximity of the pyridinium ions.![Photochemically and thermally controllable switching in the [2]rotaxane (E/Z)-7-H22+. Larger distance between the anthracene unit and the tetramide macrocycle prevents the fluorescence quenching of the anthracene unit, allowing it to fluoresce in the (E)-7-H22+ isomer. Photo-isomerization of the succinamide (E) unit to fumaramide (Z) drives the macrocycle to the Gly-Gly unit, quenching the anthracene fluorescence.](/image/article/2007/CS/b607187b/b607187b-f7.gif) | ||
| Fig. 7 Photochemically and thermally controllable switching in the [2]rotaxane (E/Z)-7-H22+. Larger distance between the anthracene unit and the tetramide macrocycle prevents the fluorescence quenching of the anthracene unit, allowing it to fluoresce in the (E)-7-H22+ isomer. Photo-isomerization of the succinamide (E) unit to fumaramide (Z) drives the macrocycle to the Gly-Gly unit, quenching the anthracene fluorescence. | ||
8. Macroscopic transport by light-driven molecular machines
The photoisomerizable bistable [2]rotaxane134 (E/Z)-8-H22+ has been prepared (Fig. 8a). It consists of a hydrophilic fumaramide (F) unit and a hydrophobic tetrafluorosuccinamide (S) unit on its dumbbell component which play the roles of two stations for the switchable tetramide-based macrocycle (M) by virtue of tunable hydrogen-bonding interactions. The macrocycle is interlocked onto the dumbbell by two terminal diphenylmethane stoppers at both ends. In the GSCC of the rotaxane (E)-8-H22+, the hydrophilic fumaramide station provides stronger hydrogen-bonding interactions with the surrounding chair-shaped macrocycle, exposing the hydrophobic S station. Irradiation of the (E)-8-H22+ isomer with 254 nm light causes E→Zphotoisomerization , producing the (Z)-8-H22+ isomer, in which the macrocycle M moves from the maleamide station to the S station, shielding the hydrophobic section of the dumbbell component. By converting the maleamide back to fumaramide thermally (115 °C) in the presence of piperidine as a base, which disrupts the hydrogen-bonding interactions between the M and S units, the macrocycle moves back to its original F station to regenerate the (E)-8-H22+ state.![(a) Photochemically and thermally induced switching of the [2]rotaxane (E/Z)-8-H22+ containing a polarophobic and a polarophilic units on its dumbbell component. (b) and (c) Photoresponsive behavior of Au{111}-surfaces functionalized with 11-MUA and molecular shuttle (E/Z)-8-H22+. (d1–d4) Lateral photographs of photo-driven transport of a 1.25 µL diiodomethane drop on a (E/Z)-8-H22+.11-MUA.Au{111} substrate on mica up a 12° incline. (Redrawn with permission from ref. 134. Copyright (2005) Nature Publishing Group.)](/image/article/2007/CS/b607187b/b607187b-f8.gif) | ||
| Fig. 8 (a) Photochemically and thermally induced switching of the [2]rotaxane (E/Z)-8-H22+ containing a polarophobic and a polarophilic units on its dumbbell component. (b) and (c) Photoresponsive behavior of Au{111}-surfaces functionalized with 11-MUA and molecular shuttle (E/Z)-8-H22+. (d1–d4) Lateral photographs of photo-driven transport of a 1.25 µL diiodomethane drop on a (E/Z)-8-H22+.11-MUA.Au{111} substrate on mica up a 12° incline. (Redrawn with permission from ref. 134. Copyright (2005) Nature Publishing Group.) | ||
The polarity of Au{111} surfaces functionalized with physisorbed monolayers of (E/Z)-9-H22+ and 11-mercaptoundecanoic acid (MUA) can be switched134 between being polarophobic (Fig. 8b) in the ground state and polarophilic (Fig. 8c) in the photo-excited state . The polarity switching depends upon the macrocycle's location on the dumbbell component. Consequently, the contact angles (θ) of small droplets (0.5–5 µL) of low-volatile polar solvents—such as, water, diiodomethane, formamide and ethylene glycol—on the (E/Z)-8-H22+-functionalized surface can be altered134 significantly (8–22°) by irradiation. For example, the diiodomethanecontact angle of 35° before UV-irradiation of the polarophobic surface (exposed S station) decreases to 13° upon UV-irradiation, a process which transforms the surface to a polarophilic one by shielding the hydrophobic S station with the macrocycle M. Similarly, θ changes for water from 55° to 45°, for formamide from 40° to 31°, and for ethylene glycol from 48° to 40°. It was observed that the (E/Z)-8-H22+ molecules are not self-assembled on the Au{111} surface in any particular orientation, and in the photostationary state (PSS), the E/Z-ratio is 50 : 50. Therefore, it can be argued that the change in surface polarity does not depend on the location of the macrocycle in every single rotaxane molecule, but on the ensemble of them, in which the overall shielding or exposure of the hydrophobic S station by the macrocycle dictates the surface polarity in a given area.
This unique light-sensitive property of the rotaxane -functionalized surface has been exploited to transport134 (Fig. 8d1–d4) a 1.25 µL diiodomethane (ρ = 3.325 g mL–1) droplet from one point to another by 1.38 mm against gravity on an α = 12° slope. In this experiment, the surface directly under the leading edge of the droplet is irradiated, a process which reduces the contact angle by θ = 22° spreading the droplet in the forward direction. When the (E/Z)-8-H22+rotaxane reaches its PSS, the irradiated section of the surface becomes more polarophilic on account of the shielding of the hydrophobic S station by the macrocycle. At the same time, the un-irradiated part of the surface remains polarophobic as a consequence of the predominance of the E-8-H22+ isomer. The difference in surface polarity in two adjacent areas moves the polar solvent diiodomethane to the more polar region against gravity at 12° gradient. Therefore, the work done against gravity by the ensemble of molecular machines is 1.2 × 10–8 J. Based on the molecular footprint of ∼3 nm2 per molecule, there are ∼2 × 1012 molecules under the elongated drop just before its transport. Considering that 40% of the rotaxane molecules on the surface have been photoisomerized at the PSS during the droplet transport, the work done by each molecule against gravity is ∼1.5 × 10–12 J, that is ∼9 kJ mol–1. The macroscopic liquid transportation by ensembles of nanoscale molecular machines may be useful for delivering reactants in lab-on-chip environments by bringing individual drops containing different reagents to perform high-precision reactions or to develop microfluidic devices.
9. PET -driven switching of a hydrogen-bonded molecular motor
The [2]rotaxane 9 is composed57 (Fig. 9) of a hydrogen-bond-donating benzylic amide macrocycle, mechanically interlocked with a dumbbell component, which is equipped with a strong hydrogen-bond-accepting unit—succinamide (succ) and a poor hydrogen-bond-accepting 3,6-di-tert-butyl-1,8-naphthalimide (ni) unit. The chair-shaped macrocycle encircles the succ unit in the neutral GSCC of the rotaxane . Photoreduction of the ni station to ni˙–radical anion by irradiation at 355 nm in the presence of an electron donor (D) 1,4-diazabicyclo[2.2.2]octane (DABCO) makes it a stronger hydrogen-bond-acceptor than the succ unit. This modification in the hydrogen-bonding abilities of the recognition sites provides a driving force for the ring to move to the ni˙–radical anion along the C-12 alkyl spacer. Charge recombination (CR) between the ni˙–radical anion and the DABCO˙+radical cation generates the neutral MSCC, which relaxes back thermally to the GSCC as the ring returns to the original succ unit. Hence, the light-induced movement of the macrocycle resembles a “power stroke” and the CR-induced return of the ring is similar to the “recovery stroke” of a piston's mechanism. This reversible photo-induced movement of components in the [2]rotaxane 9 generates57 a mechanical power of ca. 10–15 W molecule–1.![Photo-induced switching in the [2]rotaxane 9 in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO) as an external electron donor–acceptor reagent.](/image/article/2007/CS/b607187b/b607187b-f9.gif) | ||
| Fig. 9 Photo-induced switching in the [2]rotaxane 9 in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO) as an external electron donor –acceptor reagent. | ||
10. A light-driven mechanically interlocked molecular rotor
Leigh and coworkers28 have synthesized the [3]catenane 10 (Fig. 10a) in which two identical hydrogen bond donating benzylic amide macrocycles (blue and purple rings) are mechanically interlocked on to a larger macrocycle containing four hydrogen bond accepting units—namely A, B, C, and D in the sequence of decreasing hydrogen-bonding ability. In the ground state of the catenane 10, the blue and purple rings encircle the two strongest recognition units A and B, respectively, which are also designed to be photoisomerizable at uniquely different wavelengths. The hydrogen bond donating units in the large macrocycle possess gradually decreasing affinity to the smaller interlocked rings—A: a secondary amide fumaramide, B: a tertiary amide fumaramide, C: a succinic amide ester , and D: an isolated amide group which makes fewer hydrogen bonding contacts than the previous three units. A benzophenone unit was attached directly to the strongest binding fumaramide unit A to ensure its selective photoisomerization to the corresponding (Z)-olefin maleamide unit at 350 nm, whereas the tertiary fumaramide unit B can be isomerized separately to its (Z)-form at 254 nm. While the (E)-olefin fumaramide units A and B have strong affinities for the blue and purple rings, the corresponding (Z)-olefins A′ and B′ offer significantly lower affinities, allowing the interlocked rings to move away from themselves upon photoisomerization . The second of the smaller interlocked rings on the larger macrocycle also serves the purpose of a guide to the first ring by occupying the second most preferred recognition unit at any given instance, forcing the first ring to move in one overall direction—a circumrotation which is illustrated in Fig. 10b. The first photoisomerization of the A unit at 350 nm forces the blue ring to move counter-clockwise from the resulting A′ unit to the third strongest hydrogen bond donating C unit because the B unit is already occupied by the purple ring. The second photoisomerization of the B unit to generate B′ moves the purple ring counter-clockwise from the second maleamide unit B′ to the next best hydrogen bond donor D because A′ is now weaker than D, and C is already occupied by the blue ring. Thermal isomerizations (100 °C) of A′ and B′ units back to their respective (E)-olefin fumaramide forms enable the blue ring to move from the C unit to the adjacent B unit counter-clockwise and purple ring from D to the adjacent A unit clockwise—a process which introduces an impediment into the unidirectionality of the rotational motion in the [3]catenane 10 under photochemical/thermal conditions. Furthermore, this sequence of motions leaves the blue ring on the B unit and purple ring on A unit, producing a constitutional isomer of the initial state. A second series of photochemical/thermal process is required to bring the blue ring back to its original A unit and purple ring to its original B unit. In the second series, however, the blue ring moves overall clockwise and the purple ring moves overall counter-clockwise—both are in opposite directions with respect to the first series of motions. This process has been monitored by 1H NMR spectroscopy in solution only.![(a) Structural formula of the four-station [3]catenane 10. (b) (1) 350 nm, CH2Cl2, 5 min, 67%; (2) 254 nm, CH2Cl2, 20 min, 50%; (3) 100 °C, C2H2Cl4, 24 h, ∼100%. (Redrawn with permission from ref. 28. Copyright (2003) Nature Publishing Group.)](/image/article/2007/CS/b607187b/b607187b-f10.gif) | ||
| Fig. 10 (a) Structural formula of the four-station [3]catenane 10. (b) (1) 350 nm, CH2Cl2, 5 min, 67%; (2) 254 nm, CH2Cl2, 20 min, 50%; (3) 100 °C, C2H2Cl4, 24 h, ∼100%. (Redrawn with permission from ref. 28. Copyright (2003) Nature Publishing Group.) | ||
11. Light-driven supramolecular machines
Another class of nanoscale machines is based55 on a pseudorotaxane (Fig. 11) which is composed of a π-electron-accepting cyclobis(paraquat-p-phenylene) (CBPQT4+) cyclophane encircling a π-electron donating 1,5-bis-[(2-hydroxyethoxy)ethoxy]-naphthalene (BHEEN) thread. Absence of any bulky stopper at the ends of the thread component allows the macrocycle to slip on and off the thread under equilibrium conditions. However, a strong π–π charge transfer (CT) interaction, assisted by [C–H⋯O] and [C–H⋯π] interactions between the BHEEN thread and CBPQT4+ ring forms a stable BHEEN⊂CBPQT4+ pseudorotaxane in 80% yield (Ka = 2.53 × 104 M–1 in MeCN, 298 K)135 at equilibrium. Photo-induced electron transfer to the CBPQT4+ ring from an external photo-active electron donor or electrochemical reduction of the ring component destabilizes the CT interactions, inducing the decomplexation of the pseudorotaxane molecules—a process which can be monitored by measuring the BHEEN-based fluorescence intensity which is higher in the dethreaded form than it is in the pseudorotaxane.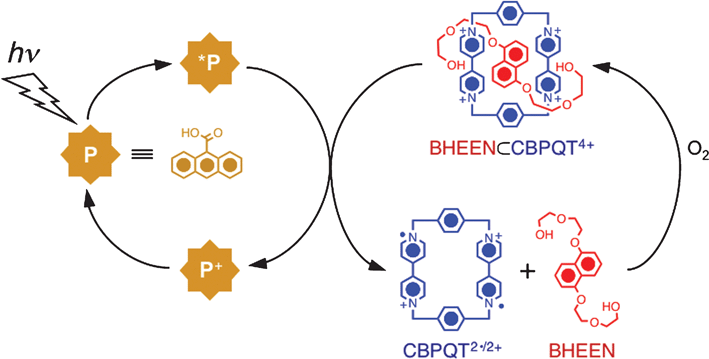 | ||
| Fig. 11 Light-induced dethreading of a BHEEN⊂CBPQT4+-based pseudorotaxane using 9-anthracenecarboxylic acid as a photosensitizer . | ||
The first generation of this series of supramolecular machines has been driven55 (Fig. 11) photochemically using 9-anthracenecarboxylic (ACA) acid as a photosensitizer (P). It donates an electron to the CBPQT4+ ring in the presence of the sacrificial reagent TEOA, thus preventing the BET process. In the absence of TEOA, BET is faster than the nuclear motion—that is, the dethreading of the ring from the thread. However, TEOA quenches the photooxidized P+ unit allowing enough time for the photoreduced CBPQT2˙/2+ to slip off the BHEEN thread, thus increasing the BHEEN-based fluorescent intensity in the 320–370 nm region. Based on the change in fluorescent intensity, 5% of the 4.8 × 10–5 M pseudorotaxane could be dethreaded after 25 min of irradiation in a deoxygenated solution in the presence of 5 × 10–6 M of P and 0.01 M TEOA. Introduction of air into the system regenerated the CBPQT4+ ring resulting in a recomplexation, a process which reproduced the original fluorescence spectrum, albeit with lower intensity.
The [2]pseudorotaxane-based supramolecular machines have been powered89 by photosensitizers, not only in solution, but also when they have been trapped physically in a rigid nanoporous sol –gel silica framework or attached covalently to silica surfaces. Irradiation of a condensed silica sol –gel matrix containing the BHEEN⊂CBPQT4+ pseudorotaxane, ACA, and a sacrificial donor , ethylenediaminetetraacetate89 (EDTA), with a 365 nm Hg-lamp (100 W) bleaches the original pink color (520 nm) of the sample arising from the CT interactions between the π-electron rich BHEEN thread and the surrounding π-accepting CBPQT4+ ring and replaces it with a pale blue color for the reduced bipyridinium units. This process is concomitant with the rise in the BHEEN-based fluorescent intensity at 320–370 nm region. Prolonged exposure of the sample to the air in the dark reproduces the original spectra, thus establishing a reversible process. These spectroscopic changes indicate that photo-induced dethreading of the pseudorotaxane occurs also in condensed phases, in which the process is slower by at least an order of magnitude than in the solution phase on account of spatial confinement within the nanopores.
In another device, the BHEEN homolog (BHEEEN) with an extra ethylene glycol chain has been attached89 covalently to a silica surface. Dipping of the BHEEEN-functionalized surface into an aqueous solution of the CBPQT4+ ring forms the corresponding pseudorotaxane, confined onto the surface, as monitored by reduced fluorescence intensity. A second immersion of the pseudorotaxane-coated surface into an ACA and EDTA solution, followed by an irradiation at 365 nm wavelength increased the naphthalene-based fluorescence intensity. Similar experiments also suggest89 that the pseudorotaxanes, attached covalently to the silica surfaces, undergo the same photo-induced dethreading in the presence of a photosensitizer and sacrificial donor .
12. Supramolecular machines powered by a light-harvesting molecular triad
A molecular triad 11 (Fig. 12)—composed of three unique electroactive components, namely, (i) an electron-donating tetrathiafulvalene (TTF) unit, (ii) a chromophoric porphyrin (P) unit, and (iii) an electron-accepting C60 unit—has been developed136,137 to harness light into electrical energy. A disulfide-based anchoring group was tagged to the TTF end of the triad in order to allow its self-assembly onto gold surfaces. When irradiated near the absorption maximum (λmax) of the chromophore P at 413 nm (40 mW cm–2 Kr-ion laser), the triad 11 undergoes a PET from the excited *P to the electron-accepting C60 unit, followed by a charge shift to the better electron donating TTF unit to generate the final charge-separated state TTF˙+–P–C60˙–. This charge-separated state is responsible for generating136,137 photocurrents in a closed electronic circuit on account of a unidirectional electron flow from the working cathode through the photoactive triad , through the electrolyte solution, to the counter electrode, and through the outer electronic circuit where the current is measured (ΔI ∼1 µA cm–2, Φphotocurrent ∼ 1%). The triad 11 has been utilized136,137 as a nanoscale power supply to drive the dethreading of the BHEEN⊂CBPQT4+ pseudorotaxane in the presence of 413 nm light at an applied potential (Vap = 0 V) that is much lower than is required for a direct electrochemical reduction (E1/2 = –300 mV) of the CBPQT4+ ring. In accordance with the PET mechanism (Fig. 12) at Vap = 0 V, the charge-separated state of the triad affords a C60˙– unit on the triad -functionalized Au-working electrode, resulting in an effective terminal potential of –550 mV which is the reduction potential of the C600/–˙ unit. This potential is high enough to reduce55,138 the CBPQT4+ ring and induce its dethreading from the BHEEN thread—a process which has been monitored by detecting the BHEEN-based fluorescence intensity. Based on the increase in the fluorescence intensity (320–370 nm), 6.7% of the 0.37 mM pseudorotaxane in MeCN could be dethreaded137 in the presence of triad -excitation over 2900 s, an estimation which is commensurate with the triad's ability to photoreduce 7% of the CBPQT4+ ring by generating 1.1 µA cm–2 photocurrent during that time. In this case, triad molecules are self-assembled onto an Au-working electrode, and so are present at a much lower concentration compared to that of the pseudorotaxanes in the medium. Furthermore, the experimental setup requires a non-air-free system contributing to the lower dethreading percentage compared to the previous system, in which the supramolecular machine, photosensitizer , and the sacrificial fuel are all present in a deoxygenated solution.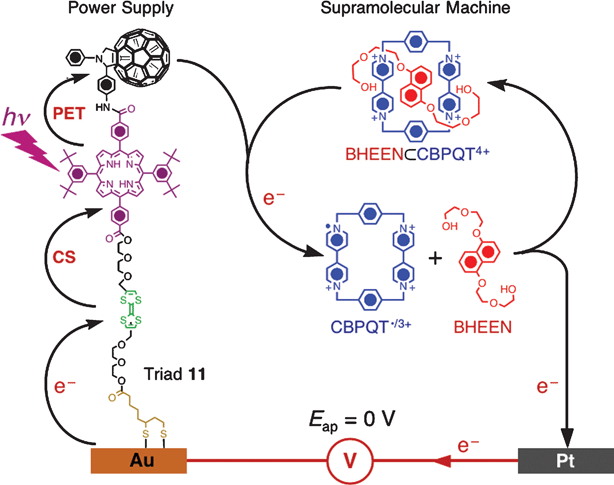 | ||
| Fig. 12 Powering of a supramolecular machine BHEEN⊂CBPQT4+ with photocurrent generated by the molecular triad 11. (Redrawn with permission from ref. 137. Copyright (2005) Wiley-VCH.) | ||
13. Light-driven unidirectional rotary motor which rotates microscale objects
Feringa and his coworkers24,27,31,35–37,139–141 have developed molecular rotary motors based on the sterically crowded, intrinsically helical-shaped molecule 12 (Fig. 13) which has a photoisomerizable elongated CC bond acting as an axle, and a stereogenic center bearing a tert-butyl substituent which adopts a pseudoaxial orientation on account of steric hindrance . The photoinduced rotary motion of such a helically chiral molecule resembles142,143 that of the ATP-synthase. The upper half of the molecule acts144 essentially as a propeller which rotates unidirectionally with respect to the bottom stator part as a result of the isomerization of the CC axle. Upon irradiation of 12 with 366 nm light, energetically uphill cis/trans isomerization of the CC bond occurs with inversion of helicity and conformational change in the cyclopentane rings directly attached to the CC bond, forcing the tert-butyl group to assume the less favored pseudoequatorial orientation. In order to regain the more favored pseudoaxial orientation, the naphthalene upper part now slips past the lower part of the molecule as a consequence of a thermal isomerization, resulting in helix inversion . A significant amount of energy is required for the thermal helical inversion to maintain a continuous unidirectional rotary motion. The unidirectional rotation of the top part of the molecule with respect to the bottom part was monitored by detecting the reversible chemical shift of the CH at the stereogenic center, which shifts up-field from 4.35 ppm (pseudoequatorial) to 4.09 ppm (pseudoaxial) upon irradiation of the stable isomer (P)-(R)-trans-12 isomer as it is converted to the unstable (M)-(R)-cis-12 isomer. Heating of the sample causes conformational changes in the five-membered ring to generate (P)-(R)-cis-12 isomer, changes which are reflected in the chemical shift of the OMe-singlet signal. Similarly, the stable cis-isomer can be converted into the original trans-isomer (P)-(R)-trans-12 by irradiating with 366 nm light, followed by a thermal flipping of the cyclopentane ring.
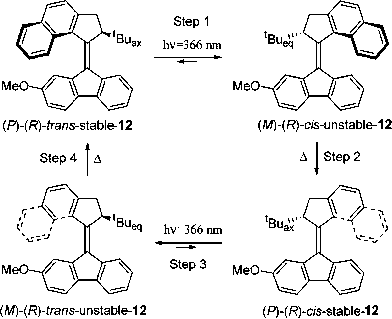 | ||
| Fig. 13 Unidirectional rotation of the helical molecular rotor 12 under photochemical and thermal conditions. (Redrawn with permission from ref. 141. Copyright (2006) American Chemical Society.) | ||
Doping144 (1 wt.%) of a similar right-handed helical alkene 13 (Fig. 14) containing a phenyl group at the stereogenic center into a liquid crystal (LC) film renders the film a polygonal texture—a characteristic of a cholesteric LC having a helix axis parallel to the surface. Irradiation of the doped LC film with 365 nm light causes a photochemical isomerization of the CC bond to occur, resulting in an inversion of helicity from the initial right-handed to the subsequent left-handed. A second inversion activated by a thermal process, followed by a second photochemical-thermal cycle, completes the full 360° rotation. Therefore, in the presence of the light the LC texture rotates clockwise whereas in the dark it is counterclockwise—a process which comes to a halt after about 10 min. The controlled texture reorganization of a molecular motor-doped LC film has been utilized144 to rotate a microscopic (5 × 28 µm) glass rod placed on the surface at an average speed of 0.67 r.p.m. clockwise photochemically and 0.22 r.p.m. counterclockwise thermally. Thus, the collective unidirectional rotary motion of an ensemble of molecular motors can be harnessed to produce mechanical movement of microscopic, and perhaps, macroscopic objects.
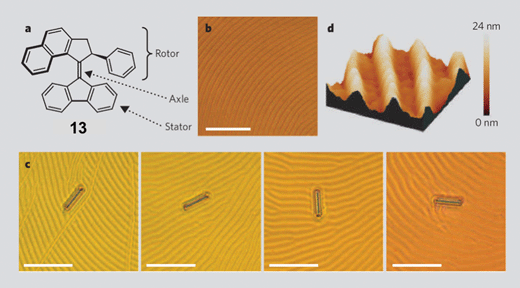 | ||
| Fig. 14 (a) Structural formula of the molecular rotor 13. (b) Polygonal texture of the liquid crystal (LC) doped with 1% of the compound 13. (c) Clockwise rotations (28°, 141°, 226°) of the glass rod (5 × 28 µm) placed on the 13-doped LC film. (d) Atomic force microscopy image (15 µm2) of the LC film. (Redrawn from ref. 144. Copyright (2006) Nature Publishing Group.) | ||
Another photoisomerizable molecular rotor 14 (Fig. 15) has been developed145 which also has a pseudoaxial methyl group at the stereogenic center. Compound (2′R)-(M)-14 can be attached to Au-surfaces via two alkylthiol pendants attached to the stator part of the molecule. The surface mounting allows the stator part to be fixed onto a stationary substrate and the propeller part can move freely on account of photochemical isomerization and thermal helix inversion to complete unidirectional 360° rotation. This process has been monitored by circular dichroism and 1H NMR spectroscopy.
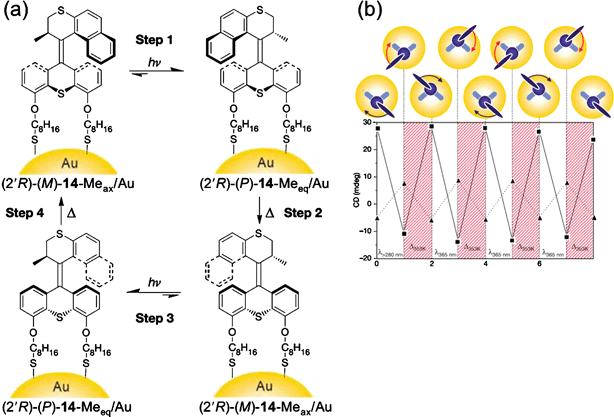 | ||
| Fig. 15 (a) Stepwise unidirectional rotation of the Au-nanoparticles functionalized with the corresponding dithiol 14 under alternating photochemical and thermal conditions. (b) Cyclic change in the CD intensity at 290 and 320 nm at each photochemical (λ > 280 nm and λ > 365 nm) and thermal (353 K) step indicating unidirectional rotation of 14. (Redrawn with permission from ref. 145. Copyright (2005) Nature Publishing Group.) | ||
14. Conclusions and outlook
In this Chapter, we have described how different noncovalent bonding interactions and controllable molecular recognition events within the mechanically interlocked bistable molecules can be executed by light as a “clean” external input. Light induces either electronic reorganizations or conformational changes in the recognition sites—a process which, in turn, alters the binding affinities of the sites with the surrounding macrocycle—triggering its relative mechanical movement within the molecule. Some of these light-driven molecular switches have been incorporated into device environments in which they switch in a manner similar to the way they do in the solution phase. When self-assembled onto surfaces, some of these bistable molecules can switch surface polarity in the presence or absence of light, a property which could be exploited in the development of microfluidic devices. Covalent attachment of bistable molecules onto the mesoporous silica surface and their light-driven mechanical switching properties have already been utilized to fabricate molecular nanovalves,146 in which the bistable rotaxane molecules act as gatekeepers. This idea could be extended to the use of these compounds for controlled release of reagents in vitro reactions or for delivery of drugs in vivo conditions. Evolution of such switchable molecules through structure–function feedback loops will help the emergence of functional molecular nanotechnology. In addition to the mechanically interlocked photo-driven molecular switches, we have discussed briefly one of the most intriguing examples of the molecular motors which exploit photoisomerization for their unidirectional rotary motion, a process that mimics the rotational motion of the ATP-synthase. When embedded into a LC film such molecular rotors can even move microscopic objects by controlling the polygonal texture of the surface.Acknowledgements
We thank all researchers—including our collaborators, colleagues, and co-workers, especially Professors Vincenzo Balzani and Alberto Credi—whose work has been presented in this Chapter for their creative contributions to the field of artificial molecular machines and devices. Our own research in this area has been supported by the Defense Advanced Research Projects Agency (DARPA), the National Science Foundation (NSF), and the Office of Naval Research (ONR).References
- H. Noji, R. Yasuda, M. Yoshida and K. Kinosita, Jr., Nature, 1997, 396, 299–302 CrossRef.
- Molecular Motors, ed. M. Schliwa, Wiley-VCH, Weinheim, 2003 Search PubMed.
- D. S. Goodsell, Bionanotechnology: Lessons from Nature, Wiley, New York, 2004 Search PubMed.
- G. Oster and H. Wang, Trends Cell Biol., 2004, 13, 114–121.
- H. Hess, G. D. Bachand and V. Vogel, Chem.–Eur. J., 2004, 10, 2110–2116 CrossRef CAS.
- R. A. L. Jones, Soft Machines: Nanotechnology and Life, Oxford University Press, Oxford, 2005 Search PubMed.
- J. F. Stoddart, Chem. Aust., 1992, 59, 576–577 and 581 CAS.
- M. Gómez-López, J. A. Preece and J. F. Stoddart, Nanotechnology, 1996, 7, 183–192 CrossRef CAS.
- V. Balzani, M. Gómez-López and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405–414 CrossRef CAS.
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS.
- A. Harada, Acc. Chem. Res., 2001, 34, 456–464 CrossRef CAS.
- C. A. Schalley, K. Beizai and F. Vögtle, Acc. Chem. Res., 2001, 34, 465–476 CrossRef CAS.
- J.-P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jiménez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS.
- R. Ballardini, V. Balzani, A. Credi, M. T. Gandolfi and M. Venturi, Struct. Bonding, 2001, 99, 163–188 CAS.
- L. Raehm and J.-P. Sauvage, Struct. Bonding, 2001, 99, 55–78 Search PubMed.
- C. A. Stainer, S. J. Alderman, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed., 2002, 41, 1769–1772 CrossRef.
- V. Balzani, A. Credi and M. Venturi, Chem.–Eur. J., 2002, 8, 5524–5532 CrossRef.
- H.-R. Tseng and J. F. Stoddart, in Modern Arene Chemistry, ed. D. Astruc, Wiley-VCH, Weinheim, 2002, p. 574–599 Search PubMed.
- V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines—A Journey into the Nano World, Wiley-VCH, Weinheim, 2003 Search PubMed.
- A. H. Flood, R. J. A. Ramirez, W.-Q. Deng, R. P. Muller, W. A. Goddard III and J. F. Stoddart, Aust. J. Chem., 2004, 57, 301–322 CrossRef CAS.
- E. R. Kay and D. A. Leigh, Top. Curr. Chem., 2005, 262, 133–177 CAS.
- A. B. Braunschweig, B. H. Northrop and J. F. Stoddart, J. Mater. Chem., 2006, 16, 32–44 RSC.
- T. R. Kelly, H. D. Silva and R. A. Silva, Nature, 1999, 401, 150–152 CrossRef CAS.
- N. Koumura, R. W. Zijlstra, R. A. van Delden, H. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS.
- Y. Yokoyama, Chem. Rev., 2000, 100, 1717–1740 CrossRef CAS.
- G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741–1754 CrossRef CAS.
- B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS.
- D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Nature, 2003, 424, 174–179 CrossRef CAS.
- S. Shinkai, M. Ikeda, A. Sugasaki and M. Takeuchi, Acc. Chem. Res., 2001, 34, 494–503 CrossRef CAS.
- K. Oh, K.-S. Jeong and J. S. Moore, Nature, 2001, 414, 889–893 CrossRef CAS.
- N. Koumura, E. M. Geertsema, M. B. van Gelder, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2002, 124, 5037–5051 CrossRef CAS.
- C. E. Godinez, G. Zepeda and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2002, 124, 4701–4707 CrossRef CAS.
- Z. Dominguez, H. Dang, M. J. Strouse and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2002, 124, 7719–7727 CrossRef CAS.
- M. F. Hawthorne, J. I. Zink, J. M. Skelton, M. J. Bayer, C. Liu, E. Livshits, R. Baer and D. Neuhauser, Science, 2004, 303, 1849–1851 CrossRef CAS.
- J. J. D. de Jong, L. N. Lucas, R. M. Kellogg, J. H. van Esch and B. L. Feringa, Science, 2004, 304, 278–281 CrossRef CAS.
- A. Koçer, M. Walko, W. Meijberg and B. L. Feringa, Science, 2005, 309, 755–758 CrossRef CAS.
- S. P. Fletcher, F. Dumur, M. M. Pollard and B. L. Feringa, Science, 2005, 310, 80–82 CrossRef CAS.
- A. S. Lane, D. A. Leigh and A. Murphy, J. Am. Chem. Soc., 1997, 119, 11092–11093 CrossRef CAS.
- P. R. Ashton, R. Ballardini, V. Balzani, I. Baxter, A. Credi, M. C. T. Fyfe, M. T. Gandolfi, M. Gómez-López, M.-V. Martínez-Díaz, A. Piersanti, N. Spencer, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 11932–11942 CrossRef CAS.
- J. W. Lee, K. Kim and K. Kim, Chem. Commun., 2001, 1042–1043 RSC.
- A. M. Elizarov, H.-S. Chiu and J. F. Stoddart, J. Org. Chem., 2002, 67, 9175–9181 CrossRef CAS.
- J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 CrossRef CAS.
- G. Kaiser, T. Jarrosson, S. Otto, Y.-F. Ng, A. D. Bond and J. K. M. Sanders, Angew. Chem., Int. Ed., 2004, 43, 1959–1962 CrossRef CAS.
- Y. Liu, A. H. Flood and J. F. Stoddart, J. Am. Chem. Soc., 2004, 126, 9150–9151 CrossRef CAS.
- J. D. Badjic, C. M. Ronconi, J. F. Stoddart, V. Balzani, S. Silvi and A. Credi, J. Am. Chem. Soc., 2006, 128, 1489–1499 CrossRef CAS.
- L. Raehm, J. M. Kern and J.-P. Sauvage, Chem.–Eur. J., 1999, 5, 3310–3317 CrossRef CAS.
- V. Bermudez, N. Capron, T. Gase, F. G. Gatti, F. Kajzar, D. A. Leigh, F. Zerbetto and S. Zhang, Nature, 2000, 406, 608–611 CrossRef CAS.
- J. M. Kern, L. Raehm, J.-P. Sauvage, B. Divisia-Blohorn and P.-L. Vida, Inorg. Chem., 2000, 39, 1555–1560 CrossRef CAS.
- R. Ballardini, V. Balzani, W. Dehaen, A. E. Dell'Erba, F. M. Raymo, J. F. Stoddart and M. Venturi, Eur. J. Org. Chem., 2000, 591–602 CrossRef CAS.
- V. Balzani, A. Credi, G. Mattersteig, O. A. Matthews, F. M. Raymo, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Org. Chem., 2000, 65, 1924–1936 CrossRef CAS.
- J.-P. Collin, J.-M. Kern, L. Raehm and J.-P. Sauvage, in Molecular Switches, ed. B. L. Feringa, Wiley-VCH: Weinheim, 2000, pp. 249–280 Search PubMed.
- A. Altieri, F. G. Gatti, E. R. Kay, D. A. Leigh, F. Paolucci, A. M. Z. Slawin and J. K. Y. Wong, J. Am. Chem. Soc., 2003, 125, 8644–8654 CrossRef CAS.
- I. Poleschak, J.-M. Kern and J.-P. Sauvage, Chem. Commun., 2004, 474–476 RSC.
- W.-Q. Deng, A. H. Flood, J. F. Stoddart and W. A. Goddard III, J. Am. Chem. Soc., 2005, 127, 15994–15995 CrossRef CAS.
- R. Ballardini, V. Balzani, M. T. Gandolfi, L. Prodi, M. Venturi, D. Philp, H. G. Ricketts and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1993, 32, 1301–1303 CrossRef.
- P. R. Ashton, R. Ballardini, V. Balzani, A. Credi, R. Dress, E. Ishow, O. Kocian, J. A. Preece, N. Spencer, J. F. Stoddart, M. Venturi and S. Wenger, Chem.–Eur. J., 2000, 6, 3558–3574 CrossRef.
- A. M. Brower, C. Frochot, F. G. Gatti, D. A. Leigh, L. Mottier, F. Paolucci, S. Roffia and G. W. H. Wurpel, Science, 2001, 291, 2124–2128 CrossRef CAS.
- J.-P. Collin, A.-C. Laemmel and J.-P. Sauvage, New J. Chem., 2001, 25, 22–24 RSC.
- G. Bottari, D. A. Leigh and E. M. Pérez, J. Am. Chem. Soc., 2003, 125, 1360–1361.
- F. G. Gatti, S. Len, J. K. Y. Wong, G. Bottari, A. Altieri, M. A. F. Morales, S. J. Teat, C. Frochot, D. A. Leigh, A. M. Brower and F. Zerbetto, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10–14 CrossRef CAS.
- A. Altieri, G. Bottari, F. Dehez, D. A. Leigh, J. K. Y. Wong and F. Zerbetto, Angew. Chem., Int. Ed., 2003, 42, 2296–2300 CrossRef CAS.
- A. M. Brower, S. M. Fazio, C. Frochot, F. G. Gatti, D. A. Leigh, J. K. Y. Wong and G. W. H. Wurpel, Pure Appl. Chem., 2003, 75, 1055–1060 CrossRef.
- V. Balzani, M. Clemente-León, A. Credi, B. Ferrer, M. Venturi, A. H. Flood and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1178–1183 CrossRef CAS.
- V. Balzani, M. Clemente-León, A. Credi, M. Semeraro, M. Venturi, H.-R. Tseng, S. Wenger, S. Saha and J. F. Stoddart, Aust. J. Chem., 2006, 59, 193–206 CrossRef CAS.
- M. Asakawa, P. R. Ashton, V. Balzani, A. Credi, C. Hammers, G. Mattersteig, M. Montalti, A. N. Shipway, N. Spencer, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 333–337 CrossRef CAS.
- V. Balzani, A. Credi, G. Mattersteig, O. A. Matthews, F. M. Raymo, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Org. Chem., 2000, 65, 1924–1936 CrossRef CAS.
- For an introduction to the early degenerate [2]catenanes that preceeded the bistable ones, see: P. R. Ashton, T. T. Goodnow, A. E. Kaifer, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1989, 28, 1396–1399 Search PubMed; P. L. Anelli, P. R. Ashton, R. Ballardini, V. Balzani, M. Delgado, M. T. Gandolfi, T. T. Goodnow, A. E. Kaifer, D. Philp, M. Pietraszkiewicz, L. Prodi, M.V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, J. Am. Chem. Soc., 1992, 114, 193–218 CrossRef.
- R. A. Bissell, E. Córdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CrossRef.
- P.-L. Anelli, M. Asakawa, P. R. Ashton, R. A. Bissell, G. Clavier, R. Górski, A. E. Kaifer, S. J. Langford, G. Mattersteig, S. Menzer, D. Philp, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, M. S. Tolley and D. J. Williams, Chem.–Eur. J., 1997, 3, 1136–1150 CrossRef CAS.
- J. O. Jeppesen, K. A. Nielsen, J. Perkins, S. A. Vignon, A. Di Fabio, R. Ballardini, M. T. Gandolfi, M. Venturi, V. Balzani, J. Becher and J. F. Stoddart, Chem.–Eur. J., 2003, 9, 2982–3007 CrossRef CAS.
- J. O. Jeppesen, S. A. Vignon and J. F. Stoddart, Chem.–Eur. J., 2003, 9, 4611–4625 CrossRef CAS.
- H.-R. Tseng, S. A. Vignon, P. C. Celestre, J. Perkins, J. O. Jeppesen, A. Di Fabio, R. Ballardini, M. T. Gandolfi, M. Venturi, V. Balzani and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 155–172 CrossRef CAS.
- B. W. Laursen, S. Nygaard, J. O. Jeppesen and J. F. Stoddart, Org. Lett., 2004, 6, 4167–4170 CrossRef CAS.
- T. Iijima, E. Apostoli, V. Balzani and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 6375–6392 CrossRef.
- J. O. Jeppesen, S. Nygaard, S. A. Vignon and J. F. Stoddart, Eur. J. Org. Chem., 2005, 196–220 CrossRef.
- For a discussion of the very first example of a degenerate [2]rotaxane that was called a molecular shuttle, see: P.-L Anelli, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1991, 113, 5131–5132 Search PubMed.
- C. P. Collier, G. Mattersteig, E. W. Wong, Y. Luo, K. Beverly, J. Sampaio, F. M. Raymo, J. F. Stoddart and J. R. Heath, Science, 2000, 289, 1172–1175 CrossRef CAS.
- A. R. Pease, J. O. Jeppesen, J. F. Stoddart, Y. Luo, C. P. Collier and J. R. Heath, Acc. Chem. Res., 2001, 34, 434–444.
- Y. Luo, C. P. Collier, J. O. Jeppesen, K. A. Neilsen, E. DeIonno, G. Ho, J. Perkins, H.-R. Tseng, T. Yamamoto, J. F. Stoddart and J. R. Heath, ChemPhysChem, 2002, 3, 519–525 CrossRef CAS.
- M. R. Diehl, D. W. Steuerman, H.-R. Tseng, S. A. Vignon, A. Star, P. C. Celestre, J. F. Stoddart and J. R. Heath, ChemPhysChem, 2003, 4, 1335–1339 CrossRef CAS.
- H.-R. Tseng, D. Wu, N. Fang, X. Zhang and J. F. Stoddart, ChemPhysChem, 2004, 5, 111–116 CrossRef CAS.
- D. W. Steuerman, H.-R. Tseng, A. J. Peters, A. H. Flood, J. O. Jeppesen, K. A. Nielsen, J. F. Stoddart and J. R. Heath, Angew. Chem., Int. Ed., 2004, 43, 6486–6491 CrossRef CAS.
- A. H. Flood, A. J. Peters, S. A. Vignon, D. W. Steuerman, H.-R. Tseng, S. Kang, J. R. Heath and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 6558–6464 CrossRef CAS.
- A. H. Flood, J. F. Stoddart, D. W. Steuerman and J. R. Heath, Science, 2004, 306, 2055–2056 CrossRef CAS.
- S. S. Jang, Y. H. Kim, W. A. Goddard III, A. H. Flood, B. W. Laursen, H.-R. Tseng, J. F. Stoddart, J. O. Jeppesen, J. W. Choi, D. W. Steuerman, E. DeIonno and J. R. Heath, J. Am. Chem. Soc., 2005, 127, 1563–1575 CrossRef CAS.
- P. M. Mendes, A. H. Flood and J. F. Stoddart, Appl. Phys. A: Mater. Sci. Process., 2005, 80, 1197–1209 CrossRef.
- N. W. P. Moonen, A. H. Flood, J. M. Fernandez and J. F. Stoddart, Top. Curr. Chem., 2005, 262, 99–132 CAS.
- J. W. Choi, A. H. Flood, D. W. Steuerman, S. Nygaard, A. B. Braunschweig, N. N. P. Moonen, B. W. Laursen, Y. Luo, E. Delonno, A. J. Peters, J. O. Jeppesen, K. Xe, J. F. Stoddart and J. R. Heath, Chem.–Eur. J., 2006, 12, 261–279 CrossRef CAS.
- S. Chia, J. Cao, J. F. Stoddart and J. I. Zink, Angew. Chem., Int. Ed., 2001, 40, 2447–2451 CrossRef CAS.
- K. Kim, W. S. Jeon, J.-K. Kang, J. W. Lee, S. Y. Jon, T. Kim and K. Kim, Angew. Chem., Int. Ed., 2003, 42, 2293–2296 CrossRef CAS.
- B. Long, K. Nikitin and D. Fitzmaurice, J. Am. Chem. Soc., 2003, 125, 15490–15498 CrossRef CAS.
- R. Hernandez, H.-R. Tseng, J. W. Wong, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2004, 126, 3370–3371 CrossRef CAS.
- T. J. Huang, H.-R. Tseng, L. Sha, W. Lu, B. Brough, A. H. Flood, B.-D. Yu, P. C. Celestre, J. P. Chang, J. F. Stoddart and C.-M. Ho, Nano Lett., 2004, 4, 2065–2071 CrossRef CAS.
- I. C. Lee, C. W. Frank, T. Yamamoto, H.-R. Tseng, A. H. Flood, J. F. Stoddart and J. O. Jeppesen, Langmuir, 2004, 20, 5809–5818 CrossRef CAS.
- T. J. Huang, B. Brough, C.-M. Ho, Y. Liu, A. H. Flood, P. A. Bonvallet, H.-R. Tseng, J. F. Stoddart, M. Baller and S. Magonov, Appl. Phys. Lett., 2004, 85, 5391–5393 CrossRef CAS.
- E. Katz, L. Sheeney-Haj-Ichia and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 3292–3300 CrossRef CAS.
- O. Lioubashevski, V. I. Chegel, F. Patolsky, E. Katz and I. Willner, J. Am. Chem. Soc., 2004, 126, 7133–7143 CrossRef CAS.
- Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, H.-R. Tseng, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. Solares, W. A. Goddard III, C.-M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745–9759 CrossRef CAS.
- T. Nguyen, H.-R. Tseng, P. C. Celestre, A. H. Flood, Y. Liu, J. I. Zink and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10029–10034 CrossRef CAS.
- K. Norgaard, B. W. Laursen, S. Nygaard, K. Kjaer, H.-R. Tseng, A. H. Flood, J. F. Stoddart and T. Bjørnholm, Angew. Chem., Int. Ed., 2005, 44, 7035–7039 CrossRef.
- D. H. Busch and N. A. Stephenson, Coord. Chem. Rev., 1990, 100, 119–154 CrossRef CAS.
- J. S. Lindsey, New J. Chem., 1991, 15, 153–180 CAS; (a) D. Philp and J. F. Stoddart, Synlett, 1991, 445–458 CrossRef CAS.
- C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303–5311 CrossRef CAS.
- S. Anderson, H. L. Anderson and J. K. M. Sanders, Acc. Chem. Res., 1993, 26, 469–475 CrossRef CAS.
- A. P. Bisson, F. J. Carver, C. A. Hunter and J. P. Waltho, J. Am. Chem. Soc., 1994, 116, 10292–10293 CrossRef CAS.
- C. A. Hunter, Angew. Chem., Int. Ed. Engl., 1995, 34, 1079–1081 CrossRef CAS.
- J. P. Schneider and J. W. Kelly, Chem. Rev., 1995, 95, 2169–2187 CrossRef CAS.
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1154–1196 CrossRef.
- A. G. Kolchinski, N. W. Alcock, R. A. Roesner and D. H. Busch, Chem. Commun., 1998, 1437–1438 RSC.
- M. Fujita, Acc. Chem. Res., 1999, 32, 53–61 CrossRef CAS.
- J. Rebek, Jr., Acc. Chem. Res., 1999, 32, 278–286 CrossRef.
- Templated Organic Synthesis, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1999 Search PubMed.
- M. Nakash, Z. C. Watson, N. Feeder, S. J. Teat and J. K. M. Sanders, Chem.–Eur. J., 2000, 6, 2112–2119 CrossRef CAS.
- J. K. M. Sanders, Pure Appl. Chem., 2000, 72, 2265–2274 CrossRef CAS.
- D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988–1011 CrossRef CAS.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS.
- J. F. Stoddart and H.-R. Tseng, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4797–4800 CrossRef CAS.
- R. L. E. Furlan, S. Otto and J. K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4801–4804 CrossRef CAS.
- D. Joester, E. Walter, M. Losson, R. Pugin, H. P. Merkle and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1486–1490 CrossRef CAS.
- L. Hogg, D. A. Leigh, P. J. Lusby, A. Morelli, S. Parsons and J. K. Y. Wong, Angew. Chem., Int. Ed., 2004, 43, 1218–1221 CAS.
- X. Zheng, M. E. Mulcahy, D. Horinek, F. Galeotti, T. F. Magnera and J. Michl, J. Am. Chem. Soc., 2004, 126, 4540–4542 CrossRef CAS.
- K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS.
- S. J. Cantrill, K. S. Chichak, A. J. Peters and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 1–9 CrossRef CAS.
- K. C.-F. Leung, F. Aricó, S. J. Cantrill and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 5808–5810 CrossRef CAS.
- F. Aricó, T. Chang, S. J. Cantrill, S. I. Khan and J. F. Stoddart, Chem.–Eur. J., 2005, 11, 4655–4666 CrossRef.
- F. Aricó, J. D. Badjic, S. J. Cantrill, A. H. Flood, K. C.-F. Leung, Y. Liu and J. F. Stoddart, Top. Curr. Chem., 2005, 249, 203–259 CAS.
- D. H. Busch, Top. Curr. Chem., 2005, 249, 1–65 CAS.
- N. Armaroli, V. Balzani, J.-P. Collin, P. Gaviña, J.-P. Sauvage and B. Ventura, J. Am. Chem. Soc., 1999, 121, 4397–4408 CrossRef CAS.
- P. Mobian, J.-M. Kern and J.-P. Sauvage, Angew. Chem., Int. Ed., 2004, 43, 2392–2395 CrossRef CAS.
- Q.-C. Wang, D.-H. Qu, J. Ren, K. Chen and H. Tian, Angew. Chem., Int. Ed., 2004, 43, 2661–2665 CrossRef CAS.
- H. Murakami, A. Kawabuchi, R. Matsumoto, T. Ido and N. Nakashima, J. Am. Chem. Soc., 2005, 127, 15891–15899 CrossRef CAS.
- E. M. Pérez, D. T. F. Dryden, D. A. Leigh, G. Teobaldi and F. Zerbetto, J. Am. Chem. Soc., 2004, 126, 12210–12211 CrossRef CAS.
- J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 704–710 CrossRef CAS.
- R. Castro, K. R. Nixon, J. D. Evanseck and A. E. Kaifer, J. Org. Chem., 1996, 61, 7298–7303 CrossRef CAS.
- S. Saha, L. E. Johansson, A. H. Flood, H.-R. Tseng, J. I. Zink and J. F. Stoddart, Small, 2005, 1, 87–90 CrossRef CAS.
- S. Saha, E. Johansson, A. H. Flood, H.-R. Tseng, J. I. Zink and J. F. Stoddart, Chem.–Eur. J., 2005, 11, 6846–6858 CrossRef CAS.
- Z. M. Liu, J. S. Yasseri, J. S. Lindsey and D. F. Bocian, Science, 2003, 302, 1543–1545 CrossRef CAS.
- R. A. van Delden, J. H. Hurenkamp and B. L. Feringa, Chem.–Eur. J., 2003, 9, 2845–2853 CrossRef CAS.
- M. K. J. ter Wiel, R. A. van Delden, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2003, 125, 15076–15086 CrossRef CAS.
- J. Vicario, M. Walko, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 5127–5135 CrossRef CAS.
- P. D. Boyer, Angew. Chem., Int. Ed., 1998, 37, 2296 CrossRef.
- J. E. Walker, Angew. Chem., Int. Ed., 1998, 37, 2308 CrossRef.
- J. Vicario, N. Katsonis, B. S. Ramon, C. W. M. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2006, 440, 163 CrossRef.
- R. A. van Delden, M. K. J. ter Wiel, M. M. Pollard, J. Vicario, N. Koumara and B. L. Feringa, Nature, 2005, 437, 1337–1339 CrossRef CAS.
- T. Nguyen, H.-R. Tseng, P. C. Celestre, A. H. Flood, Y. Liu, J. I. Zink and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10029–10034 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |