Structure and binding properties of water-soluble cavitands and capsules
Shannon M.
Biros
and
Julius
Rebek, Jr.
*
The Skaggs Institute for Chemical Biology, Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, MB26, La Jolla, CA 92037, USA. E-mail: Jrebek@scripps.edu
First published on 21st July 2006
Abstract
Synthetic receptors are modern tools for investigations into the forces involved in recognition. A widely exploited class of receptors are the resorcin[4]arene-based cavitands and capsules. This critical review (71 references) describes the evolution of water-soluble versions of these structures, along with insights the resulting host–ndash complexes have provided with regard to complexation driving forces in water. An emphasis has been placed on the influence of host structure on guest affinity and dynamics.
 Shannon M. Biros | Shannon Biros was born in Grand Haven, Michigan. She obtained a BS in chemistry from Grand Valley State University in 2001 and a PhD in chemistry from The Scripps Research Institute under the tutelage of Professor Julius Rebek, Jr. She has been awarded graduate level fellowships from The Skaggs Institute for Chemical Biology and the ARCS Foundation. Following her graduate work with Professor Rebek, she will take a postdoctoral position with Professor Kenneth N. Raymond at The University of California, Berkeley. |
 Julius Rebek, Jr. | Julius Rebek, Jr. was born in Hungary in 1944 and lived in Austria from 1945–49. He and his family then settled in the USA in Kansas. He received his undergraduate education at the University of Kansas in 1966, and obtained the PhD degree from the Massachusetts Institute of Technology (1970) for studies in peptide chemistry with Professor D. S. Kemp. As an Assistant Professor at the University of California at Los Angeles (1970–1976) he developed the three-phase test for reactive intermediates. In 1976 he moved to the University of Pittsburgh where he rose to the rank of Professor of Chemistry and developed cleft-like structures for studies in molecular recognition. In 1989 he returned to the Massachusetts Institute of Technology, where he was the Camille Dreyfus Professor of Chemistry and devised synthetic, self-replicating molecules. In July of 1996, he moved his research group to The Scripps Research Institute to become the Director of The Skaggs Institute for Chemical Biology, where he continues to work in molecular recognition and self-assembling systems. |
1. Introduction
The development of host–ndash systems that operate in water is a desirable but challenging goal for researchers working in the field of molecular recognition. While organic-soluble systems have offered insight into the forces involved in binding, particularly those affecting selectivity, they do not account for the strong desolvation and entropic benefits experienced in water. As many research groups have discovered, working in water presents many problems that are either not an issue in organic solvents or simply easier to manage. A common problem is solubility: often each piece of a multicomponent system is water-soluble, yet the resulting complex is not. Obtaining the desired protonation state of each functional group can also be troublesome. Buffers are required to maintain the appropriate pH, but depending on the buffer's identity, they can lead to more solubility problems and often change the binding properties of the host.One of the earliest accounts of molecular recognition in water involved the formation of a non-stoichiometric complex between deoxycholic acid and hydrolyzed fatty acids.1 On a historical note, this represented the first time that the terms “host” and “guest” were used to describe their roles in chemical complexation. It was not long before a series of synthetic, macrocyclic receptors with stoichiometric complexation properties followed: Pedersen's crown ethers,2,3 cyclodextrins,4 cyclophanes,5,6 calix[n]arenes7 and the glycoluril-derived cucurbiturils (Fig. 1).8 These host structures offer a significant interior concave surface available for contact with convex guests, but their large openings provided rapid on/off rates and precluded the formation of long-lived complexes. At the other end of the spectrum lie the reversibly formed metal–ndash encapsulation assemblies. The reversible interactions between metal and ligand can approach the stability of a covalent bond while still allowing the system to find a thermodynamic minimum. There have been many examples of these complexes in the literature over the past decade; these structures fall outside the scope of this review but have been discussed elsewhere.9–15
![Structures of other water-soluble macrocyclic hosts: (from left to right) [18]crown-6, cucurbit[6]uril and β-cyclodextrin.](/image/article/2007/CS/b508530f/b508530f-f1.gif) | ||
| Fig. 1 Structures of other water-soluble macrocyclic hosts: (from left to right) [18]crown-6, cucurbit[6]uril and β-cyclodextrin. | ||
The resorcin[n]arene based cavitands and capsules show behavior somewhere in between the hosts described above. The initial structures were quite flexible and consequently existed as short-lived complexes. As researchers decorated them with more and more recognition features, they observed increased binding selectivity as well as stronger host–ndash associations with slower dissociation rates. This review surveys recent developments concerning receptors that more or less completely surround their guests in water but have one open end. We have not been exhaustive in this literature survey, but in the neglected cases an effort has been made to direct the reader to the most informative publications and leading references.
2. Water-soluble cavitands
2.1 Simple cavitands
The condensation of resorcinol1 with a wide range of aldehydes provides resorcin[4]arenes 3 (Fig. 2). A few specific resorcin[4]arenes are shown as 3a–e bearing varied pendant alkyl groups on their lower rim (a.k.a. “feet”). The earliest reports of this reaction date back to the 1880's, although the product(s) could not be characterized.16–22 In 1940 Niederl and Vogel23 determined the tetrameric structure and 40 years later Högberg24–26 developed the efficient synthetic procedure still in use today.![Top: synthesis of resorcin[4]arenes bearing a variety of pendant R groups; bottom: structure of tetraanionic resorcin[4]arene host 4 and some suitable guests.](/image/article/2007/CS/b508530f/b508530f-f2.gif) | ||
| Fig. 2 Top: synthesis of resorcin[4]arenes bearing a variety of pendant R groups; bottom: structure of tetraanionic resorcin[4]arene host 4 and some suitable guests. | ||
Exposure of the simple resorcin[4]arene 3a with pendant methyl groups to excess NaOH produces the tetraanionic structure 4—where only one hydroxyl group on each aromatic ring is deprotonated.27 The resulting structure is surprisingly stable due to the formation of four strong intramolecular hydrogen bonds and the efficient delocalization of charge. This shallow bowl-shaped host binds small tetraalkylammonium salts, such as the tetramethylammonium ion and acetylcholine chloride, with association constants in the 104–105 M–1 range. Such strong binding has been attributed to favorable electrostatic interactions between host and guest; the neutral molecule tert-butyl alcohol has almost no affinity for the cavity (∼10 M–1). NICS calculations (HF/6-31G+) performed on the complex of host 4 with tetramethylammonium ion show that one methyl group is interacting with the aromatic rings of the resorcin[4]arene. This allows the thin layer of positive charge around the ammonium center to make favorable contacts with the tetraanionic upper rim. Guest exchange in this system is fast on the NMR time scale.
In order to create hosts with larger and less flexible cavities, these bowl-shaped resorcin[4]arenes were rigidified by reaction with four equivalents of bromochloromethane (Fig. 3a).28 The neighboring hydroxyl groups become bridged with a methylene spacer to give “simple cavitands”. Further modifications (i.e. attachment of solubilizing groups) can be carried out on either the pendant alkyl groups on the lower rim or by additional substitutions to the upper rim (Fig. 3b,c)29–31
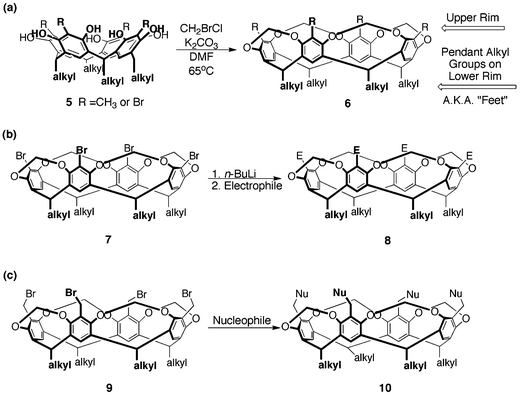 | ||
| Fig. 3 Synthesis of (a) methylene bridged cavitands and methods to derivatize the upper rim by (b) halogenation/lithiation and (c) nucleophillic substitution. | ||
![Left column: depiction of general methylene-bridged resorcin[4]arene cavitands where R = methyl (some protons and the pendant alkyl feet have been omitted for clarity); Right columns: Solubilizing groups appended to the upper rim of simple cavitands and examples of a suitable guest.](/image/article/2007/CS/b508530f/b508530f-f4.gif) | ||
| Fig. 4 Left column: depiction of general methylene-bridged resorcin[4]arene cavitands where R = methyl (some protons and the pendant alkyl feet have been omitted for clarity); Right columns: Solubilizing groups appended to the upper rim of simple cavitands and examples of a suitable guest. | ||
Hong and co-workers produced cavitand 14 by alkylation of these hydroxymethyl groups with isophthalates.34 Under basic conditions, this presumably octa-anionic cavitand binds cationic guests such as N-methylpyridinium, acetylcholine and N,N,N,4-tetramethylbenzenaminium with association constants ranging from 101 to 103 M–1. An anionic guest, sodium 4-methylbenzoate, was found to have no affinity for this host, most likely due to unfavorable electrostatic interactions.
Tetra(bromomethyl)cavitand 935,36 bearing methyl, pentyl or undecyl pendant groups was alkylated with pyridine to give cavitands 15–17 (Fig. 4).37 These tetracationic hosts presenting sp2 ammonium centers were all soluble in water, but 16 and 17 were found to aggregate. Derivative 15 with pendant methyl groups was exposed to p-cresol and p-toluenesulfonate to produce 1 ∶ 1 complexes with association constants of 1.1 × 102 and 5.2 × 102 M–1, respectively. The authors attempted 2D NMR experiments to determine the exact orientation of the guests in the resorcin[4]arene cavity, but unfortunately in–out exchange was too fast on the 1H NMR time scale.
Similar in structure to this cavitand , but presenting an sp3 ammonium center, is the tetra-hexamethylenetetramine cavitand 18.38 Extensive guest-binding studies revealed strong electrostatic contributions to guest binding with mono-anionic guests such as the sodium salt of 4-methylbenzoate. It shows an association constant of ∼102 M–1, while introduction of a second anionic center (4-methoxyisophthalate) increases the binding constant by up to two orders of magnitude. Cationic compounds, such as anilinium derivatives, have no affinity for the cavity—again due to repulsive electrostatic effects.
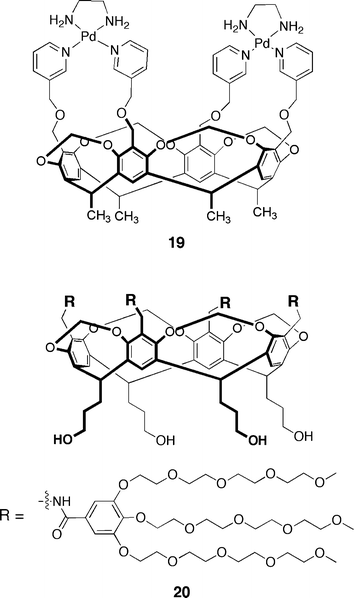
In order to achieve larger association constants, Lim and Hong prepared the Pd(II) complexed cavitand 19 shown in Fig. 5.39 Although the extended “walls” were not enough to induce slow in–out guest exchange on the NMR time scale (kinetic stability), the observed association constants were an order of magnitude greater than that for previous systems. The sodium salt of p-anisic acid bound with a Ka of 105 M–1. Other research groups have used large, neutral polyethyleneglycol dendrimers to solubilize the cavitand macrocycle (20).40 These structures bind neutral aromatics such as p-cresol, toluene and phenol with binding constants in the 104 M–1 range. Again, this increased binding affinity is attributed to the host's larger (aromatic) surface area available for contact with the guest.
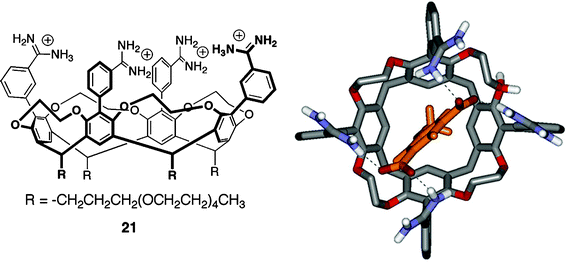 | ||
| Fig. 6 Left: Structure of Diederich's ethylene bridged cavitand 21 bearing PEG groups on the lower rim; Right: top view of energy-minimized model of cavitand 21–methoxyisophthalate complex showing hydrogen bonds between host and guest (dashed lines, some protons and the pendant chains have been omitted for clarity). | ||
In D2O, the 1 ∶ 2 host–ndash complex is likely composed of one isophthalate guest occupying the hydrophobic cavity of the cavitand while the second guest is present outside the cavity—perhaps associated by electrostatic and/or non-specific hydrophobic contacts. This binding mode allows both guest molecules to make favorable electrostatic contacts with the amidinium groups on the upper rim of the host. Molecular modeling by the authors support this hypothesis (Fig. 6). Additional binding experiments carried out in borate buffered aqueous solutions (5 mM Na2B4O7, pH 9.2) show the formation of only 1 ∶ 1 host–ndash complexes. The borate ions interact strongly with the amidinium groups of the host and effectively block any external binding sites. The Ka1 for methoxyisophthalate remained the same (within experimental error) as in unbuffered D2O, while the first association constant for nitroisophthalate was reduced by a factor of four.
In TRIS/HCl-buffered D2O, cavitand 21 also forms 1 ∶ 1 complexes with a variety of nucleotides. Complexation strength was correlated strongly with guest charge: cAMP < AMP < ADP < ATP. This series showed association constants ranging from 103 M–1 to 105 M–1. Other nucleotide monophosphates were bound (GMP, CMP, TMP, UMP), but with less affinity than the adenine derivatives. 1H NMR data revealed that the nucleotides were oriented with their aromatic ring near the cavity of host 21, allowing the phosphate(s) to interact with the amidinium groups of the upper rim.
Thermodynamic analysis of complex formation revealed that binding of both the isophthalates and nucleotides was enthalpically driven. This type of complexation was also observed by Sherman and co-workers with a simple cavitand bearing phosphate groups on the pendant alkyl chains.42,43 While the entropically favorable hydrophobic effect is contributing to complexation, the process of “creating order” among the host and guest is very unfavorable. Enthalpically speaking, there are many favorable polar and non-polar interactions between host and guest contributing to overall complex stability.
2.2 Deep cavitands
In order to create hosts with larger cavities, resorcin[4]arene 3 was condensed with electron-poor aromatic rings to give “deep cavitands”, like those shown in Fig. 7. Two water-soluble versions, 24 and 25, were synthesized by our group. Each bears an octa-amide upper rim and four ammonium centers on the pendant alkyl chains as solublilizing groups. Upon dissolution in water, these structures exist in the kite conformation as D2d velcraplex dimers44—most likely to maximize burial of lipophilic surfaces from aqueous solvent.45,46 Upon exposure to guests of suitable size and shape, the cavitands rearrange to the C4v vase conformation and form complexes where in–out guest exchange is slow on the NMR time scale. The consequences of this property in the 1H NMR spectrum are the observation of large Δδ's (≥3 ppm) for both host and guest proton resonances in the free and bound states. A significant energetic barrier between free and bound conformations results in the kinetic stability of these complexes.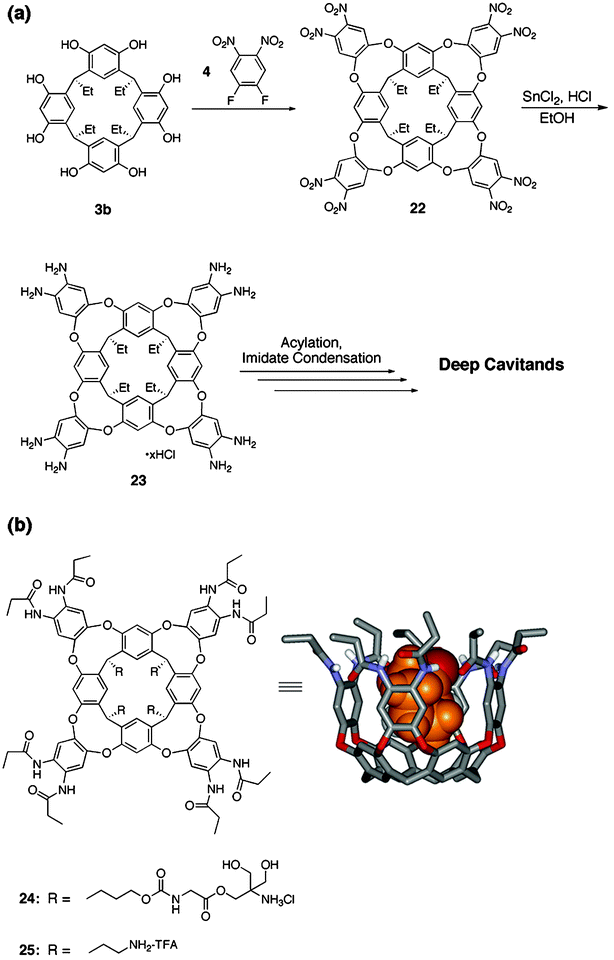 | ||
| Fig. 7 (a) General synthesis of “deep cavitands”; (b) Left: Structure of water-soluble, octaamide cavitands 24 and 25; Right: energy-minimized structure of a deep cavitand with bound cyclohexanone (some protons and the pendant alkyl groups have been omitted for clarity). | ||
A variety of suitable guests were found, most of them exhibiting a hydrophobic center bearing one polar functional group (i.e. ammonium, hydroxyl). The only guest reported to undergo fast in-out exchange on the NMR time scale was isopropyl ammonium chloride. Interestingly, this guest induced the conformational change in the host to the C4v vase shape, but was not bound tightly enough to show slow guest exchange in and out of the cavity on the NMR time scale. Guest binding affinities were modest—the maximum observed was 1.4 × 102 M–1 for the neutral cyclohexanone.
Further modification of the intermediate octaaminecavitand 23 with four carboxylate-substituted benzimidazoles gave a second water-soluble cavitand 26 (Fig. 8).47 In contrast to the previous case, this cavitand exists in the C4v symmetric vase conformation in water. The stability of this structure stems from (a) the rim of hydrogen bonds formed structure stems from (a) the rim of hydrogen bonds formed between four solvent water molecules and the nitrogen atoms of the benzimidazole rings and (b) an adventitious molecule of THF bound inside the host cavity as a remnant from the final saponification step in the preparation.
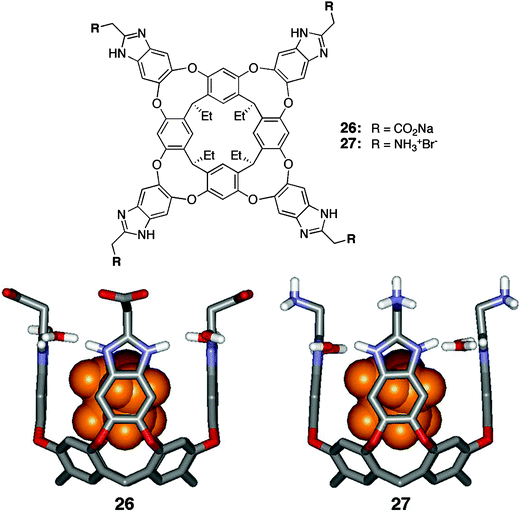 | ||
| Fig. 8 Structure of deep, tetraanionic cavitands 26 and 27 binding one molecule of THF. | ||
Some unexpected results were obtained when this cavitand was studied in solutions containing sub-micellar concentrations of sodium dodecylsulfate (SDS) or dodecyl phosphatidylcholine (DPC). Exposure of cavitand 26 to one equivalent of SDS or DPC produces a stunning host–ndash complex where the long alkyl chain appears in the host's hydrophobic cavity, and is coiled into a helix.48 This conformation fills the cavity's space with eight carbons and leaves the polar sulfate head group exposed to solvent (Fig. 9). This coiled structure was supported by the observation of NOE's between the terminal methyl group and the methylene at C-4.
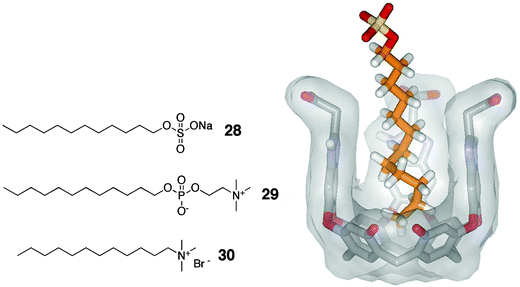 | ||
| Fig. 9 Left: Structures of SDS (28), DPC (29) and dodecyltrimethylammonium bromide (30); Right: helical SDS–cavitand 26 complex in D2O showing eight carbons in a coiled conformation. | ||
This system offers a qualitative measure of the magnitude of the hydrophobic effect. Long alkyltrimethylammonium salts present two binding sites that are known to be complementary to the host's cavity: a long alkyl chain and a trimethylammonium “knob” (choline, for example, shows a binding constant of >104 M–1 in 26 in D2O49). When dodecyltrimethylammonium bromide is added to a solution of 26 in D2O, binding of the (helical) alkyl chain, and not the trimethylammonium group is observed. The association constant is again >104 M–1. Each gauche interaction that propagates the helix conformation destabilizes the system by 0.55–0.65 kcal mol–1.50 In addition to the favorable electrostatic interactions between the tetracarboxylate upper rim and the tetraalkylammonium center, the large driving force of burying the hydrocarbon chain from water dominates the attraction.
When SDS is present in solution above its critical micellar concentration, the roles of “host” and “guest” are reversed: cavitand 26 becomes a guest inside a host structure.51 These micelles are also capable of solubilizing the cavitand in aqueous solutions when the salt concentration is too high (i.e. phosphate buffered saline).49 Cavitand 26 is still capable of binding guests under both conditions, although the resulting association constants are approximately an order of magnitude lower than in pure D2O.
This host also extracts water-insoluble species into aqueous solution. We found that n-alkanes were extracted into D2O and bound again in a helical fashion inside the cavity—just like the hydrocarbon tail of SDS. The difference in this situation is that with no polar head group present to “anchor” the orientation of the guest, the alkane tumbles rapidly on the NMR time scale inside the host's cavity.52 The protons on carbons 1 and 8 (of n-octane) experience two environments within the window of the NMR timescale. The result of this motion is an averaging of the guest proton resonances in the NMR spectra (Fig. 10).
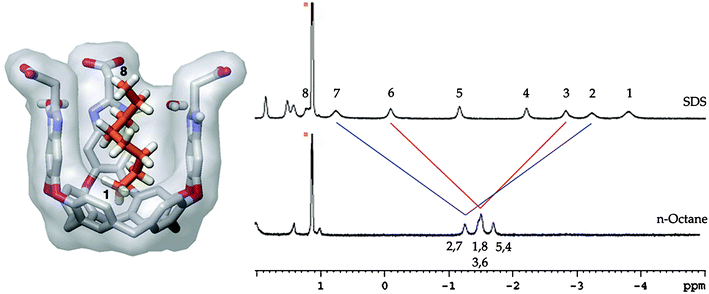 | ||
| Fig. 10 Left: model of complex between host 26 and helical n-octane; Right: Upfield region of 1H NMR spectra of SDS and n-octane in the presence of 1 mM cavitand 26; red and blue lines show “averaging” of guest resonances by fast tumbling inside the cavity. | ||
A tetracationic derivative 27 was also synthesized (Fig. 8).53 In this case, at the acidic pHs needed to obtain significant solubility in water the cavitand existed in the C2v kite conformation, without a cavity. A 25% DMSO in water solution was needed to induce the C4v vase conformation, and under these conditions guest binding was observed. This host has no affinity for cations—even tetraalkylammonium salts whose shape and size are complementary to the cavity are unable to penetrate the tetracationic upper rim of the cavitand . However, neutral and anionic adamantanes were bound in a kinetically stable fashion with association constants in the 102–103 M–1 range.
A fourth deep, water-soluble cavitand 31 bearing four benzoate groups along its upper rim is shown in Fig. 11.54 These benzoates act as “revolving doors” allowing limited access to the host's cavity. The consequences of this motion in solution become apparent in the 1H NMR spectrum of cavitand 31 in D2O. The proton resonances representing the adventitious molecule of THF bound in the cavity are sharp, in contrast to the broad peaks observed for THF bound in tetracarboxylate cavitand 26. This feature indicates the slowed in–out guest exchange rate by the rotating phenyl rings of cavitand 31.
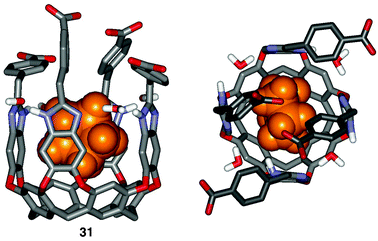 | ||
| Fig. 11 Depiction of the tetrabenzoate cavitand 31–cyclopentane complex featuring aromatic “revolving doors”. On average, two doors are suspended above the open end of the cavity at any time. | ||
The guest binding preferences of this cavitand are quite different than those observed for the tetracarboxylate cavitand 26 due to the “restricted” cavity size. While host 26 binds a wide range of n-alkanes (pentane to dodecane), tetrabenzoate cavitand 31 will only bind pentane through octane. The longer alkanes are not bound by cavitand 31. Apparently, the energy involved in disrupting the phenyl rings' positions around the rim of the cavity makes complex formation unfavorable.
3. Water-soluble molecular capsules
The creation of molecular capsules in organic solvents relies heavily on properly oriented hydrogen bonding functionalities to bring two (or more) species together in solution.55 Unfortunately, hydrogen bonds are of limited use in aqueous solvents to drive multi-component assemblies—water competes too strongly for these recognition sites. Instead, researchers in this area have relied on electrostatic interactions and the hydrophobic effect. There are relatively few examples of water-soluble molecular capsules in the literature, and we have included those associated through non-covalent and metal–ndash interactions as well as some entirely covalent analogues.3.1 Capsules assembled through non-covalent interactions
The Reinhoudt group described the preparation of calix[4]arenes derivatized on their upper rims with either amidinium, sulfonate or carboxylate groups (Fig. 12). Initial studies involving monomers 32 and 33 resulted in the precipitation of the dimeric assembly from aqueous solution, even though both monomers were water-soluble.56 (A similar result was obtained by Schrader and co-workers) with ammonium and phosphonate substituted calix[4]- and calix[6]arenes.56–58 Dissolution of the complex in methanol reveals formation of a 1 ∶ 1 heterodimeric molecular capsule containing one of the host propyl chains as a “guest”. Addition of water (up to 35%) increases the upfield shift of these protons and NOE connectivities were observed between the methyl group of this chain and the aromatic rings of the host.![Structure of (a) tetra-substituted calix[4]arene monomers; (b) depiction of a water-soluble dimeric capsule assembled through electrostatic interactions.](/image/article/2007/CS/b508530f/b508530f-f12.gif) | ||
| Fig. 12 Structure of (a) tetra-substituted calix[4]arene monomers; (b) depiction of a water-soluble dimeric capsule assembled through electrostatic interactions. | ||
Simple synthetic alterations were made to each monomer resulting in an increased water-solubility of the complex. The ethylene glycol groups of the amidinium monomer 32 were lengthened and the sulfonates of 33 were exchanged for carboxylates. Combination of calix[4]arenes 34 and 35 in a 1 ∶ 1 ratio gave the heterodimeric assembly 34·35. This complex is completely soluble in water buffered at pH 9 (Fig. 12).59 One propyl substituent is again encapsulated in the hydrophobic cavity, along with the methyl groups of the alanine amino acidic moieties. These peaks are also broadened, indicating hindered rotation of the encapsulated side chains, or intermediate rates of assembly.
Further experiments with this complex in the presence of a variety of cationic guests offered promising results. Upon exposure of capsule 34·35 to 30 equivalents of N-methylquinuclidinium, the amidinium propyl side chain resonances were shifted downfield—indicating their displacement from the cavity.60 Only one set of guest resonances was observed, suggesting fast exchange on the NMR time scale. A similar result was observed upon exposure of complex 34·35 to 10 equivalents 6-amino-2-methylquinoline: guest proton resonances were shifted upfield (max Δδ = 0.10 ppm) although fast exchange was still observed.
A second (and most spectacular) molecular capsule, formed from two deep, water-soluble cavitands was assembled via the hydrophobic effect.61 The monomers feature eight carboxylate groups which afford their solubility in water (Fig. 13). Upon exposure of this cylindrical monomer 36 to an appropriate steroidal guest, Gibb observed homodimerization to a capsule in the 1H NMR spectrum. The best guest for this dimeric system was found to be (+)-dehydroisoandrosterone 37 due to its complementary size, shape and polarity. A remarkable apparent binding constant of 1 × 108 M–1 was reported, indicating the strong influence of the hydrophobic effect. The authors cite this as the main driving force for complex formation; this interpretation is also supported by the observation of complex destruction upon addition of methanol (up to 20%).
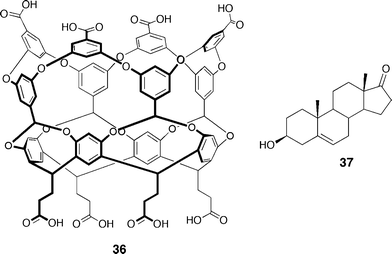 | ||
| Fig. 13 Structure of cavitand monomer 36 which dimerizes via the hydrophobic effect and structure of the best guest, (+)-dehydroisoandrosterone 37. | ||
3.2 Cage complexes: hemicarcerands
Hemicarcerands are cage-like structures assembled from two rigid bowl-shaped units, most often resorcin[3]- or resorcin[4]arenes, connected by 3–4 “bridges”.62 These bridges can range from rigid (hetero)aromatics to flexible alkyl chains. While these structures are assembled through covalent bonds, the flexible bridges create portals that allow small guests to move in and out of the cavity. The resulting hemicarceplexes are generally quite stable, and many solid-state structures have been solved.63 The most recent addition to this collection of molecules is an octahedral “nanocontainer” composed of six resorcin[4]arene units connected with ethylene diamine bridges.64 A remarkable dynamic covalent strategy was employed to achieve the synthesis of this macrostructure in one pot.The first water-soluble hemicarcerand 38 appeared in 1997 as work toward an efficient drug delivery system (Fig. 14).65 These structures have affinity for small aromatic compounds much like aspirin and acetaminophen. A justification for the cost of preparation of these “delivery agents” versus the potential payload remains dubious. Initial studies by Yoon and Cram show that this macrostructure binds small organic molecules (DMSO, EtOAc), as well as substituted aromatics such as p-xylene and 1,4-dimethoxybenzene. Complexes were formed in a 1 ∶ 1 fashion, with in–out guest exchange occurring slowly on the NMR time scale. Interestingly, tetraalkylammonium salts of suitable size were not bound by this hemicarcerand, even though they have been shown in previous systems to be complementary to the interior of a resorcin[4]arene cavity. Factors preventing this complexation could be the enthalpic desolvation costs incurred upon binding or simply size—the tetraalkylammonium salts may be too large to fit through the host's portals.
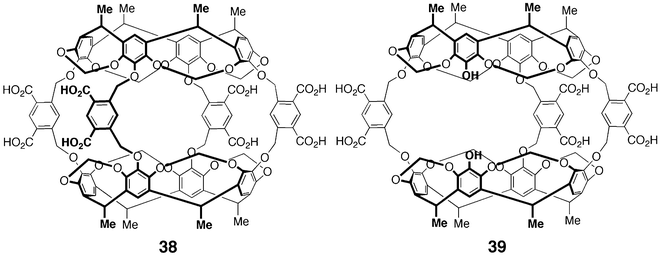 | ||
| Fig. 14 Structure of hemicarcerands 38 and 39. | ||
A closely related derivative, 39, bearing only three bridging aromatic groups was synthesized and studied by DeShayes and co-workers (Fig. 14).66 The large openings allow for in–out guest exchange to occur freely and the authors carried out an in-depth study of the complexation thermodynamics using 1H NMR and isothermal titration calorimetry (ITC). These studies estimate that for hydrophobic aromatic guests such as naphthalene, ferrocene and p-xylene, association constants are greater than 108 M–1. These complexes are stable in the solid state. Tri- and dimethoxy benzenes were all guests for the hemicarcerand, showing a strong influence of substitution pattern (guest shape) on binding affinity. Saturated cyclic structures containing hydrogen-bonding sites (i.e.camphor, norborneol) were relatively poor guests for the cavity (Ka = 103 and 104 M–1), while compounds containing charge (hexamethylenetetramine, 1-naphthoic acid) showed no affinity for the cavity of host 39. The binding properties of the deoxy-derivative were also examined and found to be identical to those of host 39.
The authors examined the thermodynamic data highlighted in the preceding paragraph and found that complexation was driven by CH-π interactions between host and guest as well as the hydrophobic effect.66 The macrocyclic effect also plays a large role in the thermodynamics of complex formation. This extensive host preorganization is the main source of the large association constants reported above. Any entropic penalties associated with organizing the host during complex formation have been paid for in covalent bonds using organic synthesis.
Analogous to the entirely covalent hemicarcerands described above, Harrison and co-workers have prepared a series of metal-bridged cage complexes able to bind small organic guests (Fig. 15).67–69 This work has been discussed previously; for more details than the description that follows we refer the reader to an earlier review.55 Functionalization of methylene-bridged resorcin[4]arene with four iminodiacetate ligands provides the scaffold for a dimeric molecular capsule, which is assembled in the presence of four metal ions [Co(II), Fe(II) or Cu(II)]. The openings are relatively small, and organic guests such as benzene and toluene cannot escape unless the capsule is disassembled.68 A crystal structure was obtained of bromobenzene bound inside the Fe(II) coordinated cage and the authors found that this guest occupied two time-averaged positions in the cavity. Both show the bromine atom near the phenyl groups, not directed at the benzylic hydrogens. It is likely this position is the best steric fit for the cavity, as well as some complementary interactions between the bromine atoms and aromatic rings.
![Structure of metal-assembled dimeric resorcin[4]arene cage 40.](/image/article/2007/CS/b508530f/b508530f-f15.gif) | ||
| Fig. 15 Structure of metal-assembled dimeric resorcin[4]arene cage 40. | ||
4. Summary and outlook
We have seen the effects of host structure on complex strength and stability through the examples described in this review. Hosts presenting multiple recognition features (ionic centers, hydrogen bonding sites) are capable of binding guests with relatively large association constants, however the in–out guest exchange rate is often fast on the NMR time scale. By rigidifying the host with intramolecular forces (hydrogen or covalent bonds) we observe a larger energetic barrier to guest exchange resulting in slower exchange rates.These systems are not meant to act as direct mimics of protein active sites and binding clefts, but as tools to study the forces involved in the recognition of small, convex molecules by a larger, concave ones. There are, however, noteworthy similarities between the cavitands and some protein hydrophobic binding sites—both are pockets lined with aromatic surfaces and decorated with hydrogen bonding sites.70,71 The capsules and cages are notional viral capsids; although there is much work to be done to create a molecular capsule made up of more than 2 subunits! At present, these structures show much promise as tools of physical organic chemistry.
Acknowledgements
We are grateful to the Skaggs Institute for Chemical Biology, the NIH and the ARCS Foundation (SMB) for funding. We also thank Dr Dariush Ajami for carrying out NICS calculations on host 4 and Janette Lundgren for assistance with manuscript preparation and helpful discussions. SMB is a Skaggs Predoctoral Fellow.References
- L. F. Fieser and M. Fieser, Steroids, Reinhold Publishing Corporation, New York, 1959 Search PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 2495–2496 CrossRef CAS.
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 CrossRef.
- Cyclodextrins, ed. J. Szejtli and T. Osa, Pergamon, Oxford, 1996, Vol. 3 Search PubMed.
- F. Diederich, Cyclophanes: Monographs in Supramolecular Chemistry, The Royal Society of Chemistry, Cambridge, UK, 1991 Search PubMed.
- Y. Murakami and O. Hayashida, in Comprehensive Supramolecular Chemistry, ed. F. Vögtle, Pergamon, Oxford, 1996, Vol. 2, pp. 419–438 Search PubMed.
- A. Casnati, D. Sciotto and G. Arena, in Calixarenes 2001, ed. Z. Asfari, Kluwer Academic Publishers, The Netherlands, 2001, pp. 440–456 Search PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS.
- D. L. Caulder and K. N. Raymond, J. Chem. Soc., Dalton Trans., 1999, 1185–1200 RSC.
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS.
- A. V. Davis, R. M. Yeh and K. N. Raymond, Proc. Natl. Acad. Sci. USA, 2002, 99, 4793–4796 CrossRef CAS.
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349–358 CrossRef CAS.
- D. W. Johnson and K. N. Raymond, Supramol. Chem., 2001, 13, 639–659 CrossRef CAS.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 371–380.
- W. J. Vickaryous, R. Herges and D. W. Johnson, Angew. Chem., Int. Ed., 2004, 43, 5831–5833 CrossRef CAS.
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1872, 5, 25 Search PubMed.
- R. Fabre, Ann. Chim. (Paris), 1922, 18, 82 Search PubMed.
- C. Liebermann and S. Lindenbaum, Ber. Dtsch. Chem. Ges., 1904, 37, 2728 CrossRef CAS.
- C. Liebermann, S. Lindenbaum and A. Glawe, Ber. Dtsch. Chem. Ges., 1904, 37, 1171 CrossRef CAS.
- E. Mertens and M. Fonteyn, Bull. Soc. Chim. Belg., 1936, 45, 186 CAS.
- A. Michael, Am. Chem. J., 1883, 5, 338 Search PubMed.
- A. Michel and J. P. Ryder, Ber. Dtsch. Chem. Ges., 1886, 19, 1388 CrossRef.
- J. B. Niederl and H. J. Vogel, J. Am. Chem. Soc., 1940, 62, 2512–2514 CrossRef CAS.
- H. Erdtman, S. Högberg, S. Abrahamsson and B. Nilsson, Tetrahedron Lett., 1968, 9, 1679–1682 CrossRef.
- A. G. S. Högberg, J. Am. Chem. Soc., 1980, 102, 6046–6050 CrossRef.
- A. G. S. Högberg, J. Org. Chem., 1980, 45, 4498–4500 CrossRef.
- H.-J. Schneider, D. Güttes and U. Schneider, Angew. Chem., Int. Ed. Engl., 1986, 25, 647–649 CrossRef.
- J. R. Moran, S. Karbach and D. J. Cram, J. Am. Chem. Soc., 1982, 104, 5826–5828 CrossRef CAS.
- H. Boerrigter, W. Verboom and D. N. Reinhoudt, Liebigs Ann./Recl., 1997, 2247–2254 Search PubMed.
- D. J. Cram, Container Molecules and Their Guests, Royal Society of Chemistry, Cambridge, 1994, Vol. 4 Search PubMed.
- J. L. Irwin and M. S. Sherburn, J. Org. Chem., 2000, 65, 602–605 CrossRef CAS.
- J. R. Fraser, B. Borecka, J. Trotter and J. C. Sherman, J. Org. Chem., 1995, 60, 1207–1213 CrossRef CAS.
- S. Pellet-Rostaing, L. Nicod, F. Chitry and M. Lemaire, Tetrahedron Lett., 1999, 40, 8793–8796 CrossRef CAS.
- S. J. Park and J.-I. Hong, Tetrahedron Lett., 2000, 41, 8311–8315 CrossRef CAS.
- T. N. Sorrell and F. C. Pigge, J. Org. Chem., 1993, 58, 784–785 CrossRef.
- K. Kim and K. Paek, Bull. Korean Chem. Soc., 1993, 14, 658 CAS.
- M. H. B. G. Gansey, F. K. G. Bakker, M. C. Feiters, H. P. M. Geurts, W. Verboom and D. N. Reinhoudt, Tetrahedron Lett., 1998, 39, 5447–5450 CrossRef CAS.
- D.-R. Ahn, T. W. Kim and J.-I. Hong, Tetrahedron Lett., 1999, 40, 6045–6048 CrossRef CAS.
- C. W. Lim and J.-I. Hong, Tetrahedron Lett., 2000, 41, 3113–3117 CrossRef CAS.
- O. Middel, W. Verboom and D. N. Reinhoudt, Eur. J. Org. Chem., 2002, 2587–2597 CrossRef CAS.
- L. Sebo and F. Diederich, Helv. Chim. Acta, 2000, 83, 93–113 CrossRef CAS.
- A. R. Mezo and J. C. Sherman, J. Org. Chem., 1998, 63, 6824–6829 CrossRef CAS.
- X. Gui and J. C. Sherman, Chem. Commun., 2001, 2680–2681 RSC.
- D. J. Cram, H.-J. Choi, J. A. Bryant and C. B. Knobler, J. Am. Chem. Soc., 1992, 114, 7748–7765 CrossRef CAS.
- T. Haino, D. M. Rudkevich and J. Rebek, Jr., J. Am. Chem. Soc., 1999, 121, 11253–11254 CrossRef CAS.
- T. Haino, D. M. Rudkevich, A. Shivanyuk, K. Rissanen and J. Rebek, Jr., Chem.–Eur. J., 2000, 6, 3797–3805 CrossRef CAS.
- F. Hof, L. Trembleau, E. C. Ullrich and J. Rebek, Jr., Angew. Chem., Int. Ed., 2003, 42, 3150–3153 CrossRef.
- L. Trembleau and J. Rebek, Jr., Science, 2003, 301, 1219–1220 CrossRef CAS.
- S. M. Biros, E. C. Ullrich, F. Hof, L. Trembleau and J. Rebek, Jr., J. Am. Chem. Soc., 2004, 126, 2870–2876 CrossRef CAS.
- E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, Wiley, New York, 1994 Search PubMed.
- L. Trembleau and J. Rebek, Jr., Chem. Commun., 2004, 58–59 RSC.
- R. J. Hooley, S. M. Biros and J. Rebek, Jr., Chem. Commun., 2006, 509–510 RSC.
- C. H. Haas, S. M. Biros and J. Rebek, Jr., Chem. Commun., 2005, 6044–6045 RSC.
- R. J. Hooley, H. J. Van Anda and J. Rebek, Jr., J. Am. Chem. Soc., 2006, 128, 3894–3895 CrossRef CAS.
- F. Hof, S. L. Craig, C. Nuckolls and J. Rebek, Jr., Angew. Chem., Int. Ed., 2002, 41, 1488–1508 CrossRef CAS.
- F. Corbellini, R. Flammengo, P. Timmerman, M. Crego-Calama, K. Versluis, A. J. R. Heck, I. Luyten and D. N. Reinhoudt, J. Am. Chem. Soc., 2002, 124, 6569–6575 CrossRef CAS.
- R. Zadmard, M. Junkers, T. Schrader, T. Grawe and A. Kraft, J. Org. Chem., 2003, 68, 6511–6521 CrossRef CAS.
- R. Zadmard, T. Schrader, T. Grawe and A. Kraft, Org. Lett., 2002, 4, 1687–1690 CrossRef CAS.
- F. Corbellini, L. D. Costanzo, M. Crego-Calama, S. Geremia and D. N. Reinhoudt, J. Am. Chem. Soc., 2003, 125, 9946–9947 CrossRef CAS.
- F. Corbellini, R. M. A. Knegtel, P. D. J. Grootenhuis, M. Crego-Calama and D. N. Reinhoudt, Chem.–Eur. J., 2005, 11, 298–307 CrossRef.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2004, 126, 11408–11409 CrossRef CAS.
- E. Maverick and D. J. Cram, in Comprehensive Supramolecular Chemistry, ed. F. Vögtle, Pergamon, Oxford, 1996, Vol. 2, pp. 367–418 Search PubMed.
- For selected examples, please see: (a) D. J. Cram, R. C. Helgeson, C. B. Knobler and E. F. Maverick, Tetrahedron Lett., 2000, 41, 9465–9470 CrossRef CAS; (b) R. C. Helgeson, C. B. Knobler and D. J. Cram, J. Am. Chem. Soc., 1997, 119, 3229–3244 CrossRef CAS; (c) Y.-S. Byun, T. A. Robbins, C. B. Knobler and D. J. Cram, J. Chem. Soc., Chem. Commun., 1995, 1947–1948 RSC; (d) M. E. Tanner, C. B. Knobler and D. J. Cram, J. Am. Chem. Soc., 1990, 112, 1659–1660 CrossRef CAS.
- X. Liu, Y. Liu, G. Li and R. Warmuth, Angew. Chem., Int. Ed., 2006, 45, 901–904 CrossRef CAS.
- J. Yoon and D. J. Cram, Chem. Commun., 1997, 497–498 RSC.
- E. L. Piatnitski, R. A. Flowers, II. and K. Deshayes, Chem.–Eur. J., 2000, 6, 999–1006 CrossRef CAS.
- O. D. Fox, N. K. Dalley and R. G. Harrison, J. Am. Chem. Soc., 1998, 120, 7111–7112 CrossRef CAS.
- O. D. Fox, N. K. Dalley and R. G. Harrison, Inorg. Chem., 1999, 38, 5860–5863 CrossRef CAS.
- O. D. Fox, J. F.-Y. Leung, J. M. Hunter, N. K. Dalley and R. G. Harrison, Inorg. Chem., 2000, 39, 783–790 CrossRef CAS.
- S. A. Jacobs and S. Khorasanizadeh, Science, 2002, 295, 2080–2083 CrossRef CAS.
- J. L. Sussman, M. Harel, F. Frolow, C. Oefner, A. Goldman, L. Toker and I. Silman, Science, 1991, 253, 872–879 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |