Transition metal-mediated P–C/X exchange at bound phosphine ligands (X = aryl, alkyl, NR2, OR and F): scope and mechanisms
Stuart A.
Macgregor
School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, UK EH14 4AS. E-mail: s.a.macgregor@hw.ac.uk
First published on 27th September 2006
Abstract
The range of transition metal-mediated P–C/X exchange reactions that result in the replacement of a phosphine substituent with another group, X, are categorised according to the nature of the replacing group (X = aryl or alkyl, N- and O-based species and fluoride). Proposed mechanisms for P–C/X exchange are described and the factors promoting these unusual—and often undesirable—reactions are discussed. This tutorial review should be of relevance for those engaged in homogeneous catalysis, C–F activation and the synthesis of complexes combining soft metal centres and hard donor ligands.
 Stuart Macgregor | Stuart Macgregor received a PhD in metallaborane chemistry from the University of Edinburgh in 1992, studying under the supervision of Professors Alan Welch and Lesley Yellowlees. From 1992–1994 he held a NATO Western European Fellowship at the Université de Paris-Sud, working with Professor Odile Eisenstein. After two years as a post-doctoral fellow at the Australian National University, Canberra, he returned to Edinburgh in 1997 to take up a lectureship in inorganic chemistry at Heriot-Watt University where he is currently a Reader. His research interests are in the application of computational chemistry to the structure and reactivity of transition metal systems. |
1. Introduction and scope of review
This review will survey reactions of transition metal–phosphine complexes, [LnTM–PR3], that result in P–C bond cleavage and the replacement of a substituent, R, by some other group, X. The substituents on phosphorus are generally simple aryl or alkyl groups, while the replacing group, X, will be another C-based moiety, an N- or O-based group, or fluoride. These processes result in a net P–C/X exchange reaction, although this is sometimes masked by the subsequent reactivity of the initial exchange product. Aspects of the activation of P–C bonds have been reviewed previously by Garrou in 1985.1 At this time the majority of examples featured the one-way transfer of substituents from phosphorus to metal, often with the formation of phosphido-bridged dimers and higher-nuclearity species. Examples of P–C/X exchange reactions that did appear in that work are also included here. During the preparation of this review Parkins has also published a survey of reactions involving the migration and cleavage of substituents from donor atoms in transition metal compounds.2The replacement of a substituent on a phosphine has important consequences for its use in metal-mediated synthesis and homogeneous catalysis. These rely on phosphine ligands acting as essentially innocent ‘spectators’, whilst conferring a specific electronic, steric or even chiral environment on the metal centre. Substituent replacement is therefore usually highly undesirable and the decomposition of homogeneous catalysts has been discussed in this context.3 The present review of P–C/X exchange reactions is organised according to the nature of the replacing group, starting with P–C/C exchange processes where most detailed studies are available. Emphasis is placed on the mechanisms by which P–C/X exchange may occur and a number of general processes have been put forward (see Fig. 1). In Mechanism A exchange proceeds via initial transfer of an R group from P to M in what is a formal oxidative addition. P–C/X exchange is completed by transfer of X from M to P, formally reductive elimination. An alternative process implicated in many cases is nucleophilic attack by X, either externally, Mechanism B, or internally, Mechanism C. Mechanistic details on these processes are scarce and, in principle, exchange could occur via a single concerted step. However, in many cases intermediates have been proposed, such as phosphonium salts formed via P–X reductive elimination, Mechanism D, or metallophosphoranes, Mechanism E. It should be noted that nucleophilic attack of hard O-based4,5 and fluoride ligands6 at transition metal-bound phosphines can also result in disproportionation, yielding low-valent metal species and phosphoranes. These processes, which are often important in catalyst activation steps, are closely related to Mechanisms B and C, but space limitations preclude their further discussion here.
 | ||
| Fig. 1 Possible mechanisms of P–C/X exchange reactions. | ||
2. P–C/C exchange reactions
P–C cleavage reactions of transition metal–phosphine complexes have been studied extensively, especially in the context of catalyst stability.1,3 The formation of a range of organic species in these studies (e.g. for PPh3 complexes, benzene, biphenyl and related coupling products) is taken as an indication that P–C bond cleavage is occurring and in many cases this is confirmed in the metal-containing product which is often a bi- or higher-nuclearity system featuring bridging phosphido groups. Such reactions have often been observed under the high temperatures and pressures relevant for the catalytic process under study (e.g. for hydroformylation with [Co2(CO)8], 190 °C and 2000 psi CO–H27). P–C/C exchange processes were also observed in some of these early studies. However, more recently it has become apparent that such processes may also occur under extremely mild conditions and in some instances below room temperature. The range of P–C/C exchange processes now available in the literature is covered below, firstly for the more common P–aryl/aryl exchange and then for P–aryl/alkyl exchange.2.1. P–aryl/aryl exchange
P–aryl/aryl exchange occurs when the alkene hydroformylation catalyst, [Rh(H)(CO)(PPh3)3], is heated to 130 °C in the presence of P(p-tolyl)3, the exchange process manifesting itself in the formation of mixed phosphines.8 This Ph/p-tolyl exchange is catalytic and a mechanism based on P–C oxidative addition and exchange via phosphido-bridged dimers such as 1 was proposed (cf. Mechanism A, Fig. 1).
A range of Rh species was shown to effect the same Ph/p-tolyl exchange process, including [RhCl(PPh3)3], [Rh(acac)(CO)(PPh3)] and [RhCl(CO)(PPh3)2], while higher temperatures were required for polynuclear Rh species, possibly reflecting the need to break these species into monomeric components. P–Ph/p-tolyl exchange is also not limited to Rh, it being observed with [Co2(CO)8], [M3(CO)12] (M = Ru, Os), [Ni(CO)2(PPh3)2] and [Pd(PPh3)4]. Other work has identified similar processes catalysed by [Pd(OAc)2].9 Further studies on the reaction of [Co2(CO)8] with PPh3–PAr3 mixtures indicated that exchange was accelerated by electron withdrawing substituents on the Ar group.7 This was also interpreted in terms of a mechanism involving initial P–C bond activation via an oxidative addition process, similar to that seen for aryl halides where the metal centre acts as a nucleophile at the ipso carbon.
The first example of direct P–aryl/aryl exchange where both reactant and product were well-defined metal complexes was reported by Kong and Cheng in 1991.10 Heating trans-[Pd(Ar)I(PPh3)2] species (Ar = C6H4–p-X; X = Me, OMe) in THF at 60 °C results in the smooth formation of the exchange products trans-[PdI(Ph)(PPh3)2] and trans-[PdI(Ph)(PPh2Ar)2], as monitored using 1H NMR spectroscopy. These species are thought to arise from the assumed initial product of exchange, trans-[PdI(Ph)(PPh2Ar)(PPh3)], via rapid intermolecular phosphine scrambling (Fig. 2). For X = Me, a 90 : 10 ratio of exchanged to non-exchanged product was seen, while this increases to 96 : 4 for X = OMe, indicating that electron donating substituents favour the exchange products. Evidence for a second Ph/Ar exchange was seen in the formation of complexes containing PPhAr2 ligands. Labelling studies indicated that a degenerate exchange process would also be expected to occur in the all-phenyl complex trans-[PdI(Ph)(PPh3)2]. Ph/Ar exchange is almost completely shut down, however, by the addition of excess PPh3 and this inverse phosphine concentration dependence turns out to be a common feature of most P–C/C exchange reactions. This observation was again interpreted in terms of a P–C oxidative addition mechanism requiring the formation of a 3-coordinate Pd centre, although this has since been questioned (see below).
![P–Ph/Ar exchange in trans-[Pd(Ar)I(PPh3)2] (Ar = C6H4–p-X; X = Me, OMe).10](/image/article/2007/CS/b516248n/b516248n-f2.gif) | ||
| Fig. 2 P–Ph/Ar exchange in trans-[Pd(Ar)I(PPh3)2] (Ar = C6H4–p-X; X = Me, OMe).10 | ||
Alternative mechanisms for Ph/Ar exchange in [Pd(Ar)X(PPh3)2] species have originated in the observation of unexpected (and unwanted) Ph-containing by-products in Pd-catalysed cross-coupling reactions. For example, Segelstein and co-workers noted that the Stille coupling reaction (eqn (1)) was dominated by the side product 3, derived from Ph exchange with the electron-rich C6H4–p-OMe group.11
 | (1) |
Product 3 could also be obtained from the reaction of p-bromoanisole with [PdCl2(MeCN)2] in the presence of [PPh4]Br, prompting the authors to propose phosphonium halides as intermediates in P–aryl/aryl exchange. These could be formed from [Pd(Ar)X(PPh3)2] via P–C reductive elimination (cf. Mechanism D, Fig. 1), with Ph/Ar exchange being completed by the oxidative addition of the phosphonium cation to Pd(0) with activation of the P–Ph bond (see Fig. 3). Similar conclusions were reached by Yamamoto and co-workers, who noted a particular tendency for trans-[Pd(Ph)X(PPh3)2] to produce phosphonium salts in chlorinated solvents.12 In addition, this paper describes the use of phosphonium salts as an aryl group source for Heck-style coupling and this approach has since been exploited in synthesis. One novel example is the formation of phosphonium salts containing up to three zinc phthalocyanine (ZnPc) moieties, [PPh(ZnPc)3]I, where Ph/ZnPc exchange appeared to be facilitated by the electron-rich nature of the ZnPc group.13
![P–Ph/Ar exchange via phosphine loss and phosphonium salt formation (X = halide).14 In low polarity media a tight ion pair, [ArPh3P]+[PdX]−, has been proposed for the boxed intermediate.15](/image/article/2007/CS/b516248n/b516248n-f3.gif) | ||
| Fig. 3 P–Ph/Ar exchange via phosphine loss and phosphonium salt formation (X = halide).14 In low polarity media a tight ion pair, [ArPh3P]+[PdX]−, has been proposed for the boxed intermediate.15 | ||
The initial reports on P–aryl/aryl exchange prompted more detailed mechanistic studies on this process. Novak and co-workers studied the reactions of a range of [Pd(Ar′)I(PAr3)2] species.14 In addition to the inverse dependence of the rate of Ar/Ar′ exchange on phosphine concentration, a similar inhibitory effect for added iodide was noted in THF, suggesting a pathway involving initial iodide dissociation. This is also consistent with enhanced exchange in more polar media. A contribution from a pathway involving direct loss of [PAr3Ar′]I from 4-coordinate [Pd(Ar′)I(PAr3)2] could not be ruled out, however. Equilibria studies indicated that exchange is promoted by electron donating groups on both the Pd- and P-bound aryl groups, but is inhibited by bulkier phosphines. [Pd{P(o-tolyl)3}2] was therefore identified as a particularly useful catalyst for cross-coupling reactions as it should minimise P–Ar/Ar′ exchange. The rate enhancement for exchange seen here with electron-rich PAr3 contrasts with the trend reported by Dubois and Garrou7 for the [Co2(CO)8]–PPh3/PAr3 system, suggesting that a different mechanism is indeed operative in that case.
Grushin extended this work to consider the role of the anionic ligand, X, in [Pd(C6D5)X(PPh3)2] species (X = I, Br, Cl, F).15 For all species, P–aryl/aryl exchange was observed in benzene at 75 °C. As Pd–X heterolysis is unlikely under these conditions, it was concluded that halide dissociation is not a prerequisite for the exchange to occur. The reaction was fastest for X = I, with relative rates of 100 : 4 : 1 for X = I, Br and Cl respectively. These data were interpreted in terms of the ease of phosphine dissociation (greatest for X = I) and a possible π-stabilisation of the unsaturated intermediate formed prior to P–C reductive elimination (greatest for X = Cl). In the low polarity media employed in this study, tight ion pairs, [Ph4P]+[PdX]−, were proposed as intermediates, rather than fully solvent-separated phosphonium salts. The propensity for phosphonium salt formation in chlorinated solvents, noted by Yamamoto,12 was rationalised in terms of facile halide loss from [Ph4P]+[PdX]− in these media. The further reaction of the highly unsaturated Pd(0) species formed with solvent C–Cl bonds would then drive the equilibrium towards phosphonium salt formation. For [Pd(C6D5)F(PPh3)2], a similar exchange mechanism is proposed but the system is complicated by additional decomposition pathways that produce unsaturated Pd(0) species. As these themselves promote PPh3 dissociation from [Pd(C6D5)F(PPh3)2], an autocatalytic effect is seen, with the result that P–aryl/aryl exchange is slightly faster than for X = Cl.
2.2. P–aryl/aryl exchange in metal-mediated synthesis
The intermediacy of complexes of the type trans-[Pd(Ar′)X(PAr3)2] in many Pd-mediated cross-coupling reactions means that P–aryl/aryl exchange is an important factor in the formation of unwanted side products. In addition to the examples described above,11–15 similar difficulties have been encountered in the Heck vinylation of aryl halides,16 Suzuki17 and Sonogashira18 couplings and C–heteroatom bond formation.19 Consistent with the above mechanistic studies, it has been found that P–aryl/aryl exchange usually becomes more problematic with electron-rich Pd–aryl groups, especially in more polar solvents. The use of bulky phosphines such as P(o-tolyl)316 or P(C6H4–o-OMe)317 has met with some success in reducing this problem, although this may be due to these ligands producing alternative pathways16 rather than reducing exchange via a steric effect as such. Above all, the variables that control P–aryl/aryl exchange will also affect catalytic activity and so it is extremely difficult to provide general strategies that will take account of both these issues.2.3 P–aryl/alkyl exchange
As with P–aryl/aryl exchange, early studies identified P–aryl/alkyl exchange through the formation of substituted phosphine ligands. PPh2(C3H7) and PPh(C3H7)2 were produced from [Rh(H)(CO)(PPh3)3] under conditions for propene hydroformylation or (more readily) hydrogenation (105 °C, 30 psi C3H6, 100 psi H2).20 These exchanged phosphines are thought to arise from oxidative addition of a P–Ph bond and coupling of the resultant PPh2 ligand with a propyl ligand, itself formed via insertion of propene into a Rh–H bond. The same exchange process occurs at a number of Rh complexes, although, interestingly, not with [RhCl(PPh3)3] or [Rh(Cl)(CO)(PPh3)2]. The fact that these latter two species do effect P–aryl/aryl exchange (see above)8 suggests that P–aryl/alkyl exchange is intrinsically the harder process. This is certainly consistent with the result of calculations on [Pd(H)3(PH2R)]− species (R = Me, Ph), where much lower barriers for Ph transfer were deduced.21 The formation of PPh2(hexyl) from the reaction of [Co2(CO)8]–PPh3 with 1-hexene under hydroformylation conditions has also been observed, presumably via an analogous mechanism to that suggested for the preceding Rh example.7In contrast to the relatively high temperatures and pressures of the above studies, P–aryl/alkyl exchange has also been observed in the room temperature reaction of [NiCl2(PPh3)2] with MeMgBr.22 This leads to the rapid formation of a range of organic coupling products as well as free PMe2Ph and PMePh2. The final metal-containing product was not defined, although the intermediacy of a metallophosphorane (Mechanism E, Fig. 1) was proposed. Similarly, the decomposition of [CoMeL3] species (L = PPh3−nMen; n = 0–2) occurs readily at 0 °C to give organic coupling products as well as Ph/Me-scrambled phosphines.23
As with P–aryl/aryl exchange, P–aryl/alkyl exchange can play a deleterious role in the formation of unwanted side products in Pd-catalysed coupling reactions. In the Stille coupling reactions between trans-[PdI(Me)(PPh3)2] and various tin reagents, contamination with arylated products arising from Ph/Me exchange was observed (e.g.eqn (2)).24
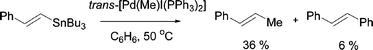 | (2) |
A detailed mechanistic study of the P–aryl/alkyl exchange reaction of trans-[PdI(Me)(PPh3)2] in benzene at 75 °C showed Ph/Me exchange to be irreversible, indicating a significant preference for the formation of P–Me and Pd–Ph bonds over P–Ph and Pd–Me bonds. This is in contrast to P–aryl/aryl exchange where equilibria are usually observed. Moreover, the P–Ph/Me exchange process does not appear to require PPh3 dissociation, nor does it involve (at least in CD2Cl2) the formation of a free phosphonium salt. These differences prompted Novak et al. to suggest that P–Ph/Me exchange in this system may proceed via an oxidative addition–reductive elimination pathway (cf. Mechanism A, Fig. 1).14 However, the formation of a free phosphonium salt is not necessarily required for exchange, as in low polarity media a tight ion pair is a feasible aryl/aryl exchange intermediate.15 The lack of dependence on phosphine concentration remains puzzling, however, and clearly further studies are required before these subtle differences are fully understood.
3. P–C/NR2 exchange reactions
In contrast to the relatively large number of P–C/C exchange reactions and their obvious implications for catalysis, very few P–C/N exchange processes have been characterised and known examples are rather esoteric in nature. Vierling and Riess found that deprotonation of the P,N-chelating phosphine in 4 led to formation of a bicyclic phosphine and displacement of a phenyl group onto iron, 6.25 Performing the deprotonation at −80 °C allowed a metallophosphorane intermediate, 5, to be characterised, thus providing an excellent model reaction for P–C/X exchange processes via Mechanism E of Fig. 1, where in this case the nucleophile is formed in situ within the phosphine ligand. Species 4 can be re-formed from 6 by protonation. A similar exchange process was observed with an allyl, rather than an aryl, group at phosphorus, although in this case a subsequent rearrangement to give an Fe–vinyl product was seen and the process proved irreversible.
The only other example of P–C/N exchange was reported by Shaw et al. and also involves an intraligand attack.26 Treatment of complex 7 with hydrazine hydrate is thought to produce intermediate 8. The terminal NH2 group of 8 then acts as a nucleophile, attacking phosphorus and inducing P–C bond cleavage. A subsequent proton transfer results in the ring expanded product 9.
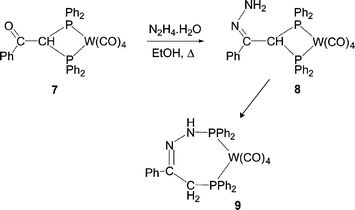
4. P–C/OR exchange reactions
Many examples of P–C/O exchange have now been observed, although taken as a whole these present a rather disparate array of reactions. In the following, an attempt is made to categorise these processes in terms of the origin of the O-based moiety. It should be stressed, however, that in the absence of mechanistic details the original and ‘active’ forms of the O-based species may not be the same. For example, O-based ligands may react as intramolecular nucleophiles or dissociate and act as external nucleophiles (Mechanisms B and C, Fig. 1). Likewise, a solvent molecule may attack phosphine directly or may bind to a metal centre prior to an intramolecular attack. In general, such nucleophilic attack seems to be considered far more prevalent in P–C/O exchange reactions than the formation of phosphonium salts that was common in P–C/C exchange.4.1 OR derived from a ligand-based carbonyl group
Shaw et al. have observed another ring expansion reaction involving the acylated dppm ligand in 7 to yield, in this case, the product 10. This process appears analogous to that observed with hydrazine hydrate (7–9 above), although in this case reaction is reported to occur only upon exposure to light.27
The bidentate ligand in 10 has also been reported by Braunstein et al., albeit arrived at by a very different route.28 In this case, the carbonyl group of one (diphenylphosphino)acetophenone in 11 is able to attack a neighbouring phosphine ligand with displacement of [PhC(O)CH2]−. This goes on to deprotonate the activated CH2 group of the bidentate ligand to form 12 and acetophenone.

Another P–C/O exchange process involving a ligand-based carbonyl moiety was observed in the reaction of [IrCl(PPh3)3] with dibenzoyldiazomethane in THF to give 13 as a minor product.29 The authors suggest that N2 loss results in the formation of a carbene intermediate which subsequently undergoes orthometallation. P–C/O exchange must then occur, with the phenyl group presumably being lost from the system as benzene.

4.2 OR derived from water or hydroxide
P–C/O exchange induced by water or hydroxide ions results in complexes of phosphinite ligands, R2P(OH), or, depending upon the reaction conditions, deprotonation may occur to give the diphenylphosphine oxide anion. An early example of water-induced P–C/O exchange was seen in [MCl2{PPh2(CCCF3)}2] (14, M = Pd, Pt) complexes, where the P–C(alkynyl) bonds break to give chloro-bridged dimers (15).30 The hydrolysis of these P–C(sp) bonds appears to be much more facile than that of analogous P–C(sp2) or P–C(sp3) bonds, with which no reaction is observed under similar conditions.31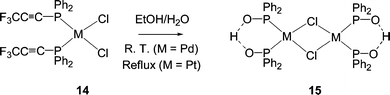
The P–C(sp3) bond of the dppm ligand can be susceptible to cleavage, however, and this is observed upon treatment of [PtCl2(dppm)] with excess NaOH in liquid ammonia.32 An initially-formed bridging amido species, 16, becomes subject to nucleophilic attack by OH−, with displacement of the P–C bond. Proton transfer completes the formation of the observed product, 17, for which both cis and trans isomers were characterised.

A similar hydrolysis of [PtCl2(dppm)] was subsequently observed under phase transfer conditions to give compound 18.33
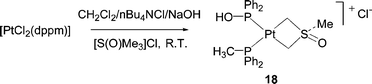
In general, P–C/X exchange reactions involving P–C(sp3) bonds are rare and the observation of this process in these dppm complexes may reflect the strain inherent in these systems. Another example where this may be a key factor is the hydroxide-assisted cleavage of a P–C bond in iminophosphorane-phosphine complexes, 19. In this case, a 3-membered ring is cleaved to give diphenylphosphine oxide complexes, 20.34

A final example of P–C/O exchange involving water differs from those above as the phosphorus centre involved is not initially bound to the metal centre (see Fig. 4).35 In [Mo(Ph2P(CH2)nPPh2)(PPh2R)2] (21, R = Me, Ph; n = 1–3) complexes, steric encumbrance is thought to induce the unusual η6-coordination mode of one PPh2R ligand. The room temperature reaction of 21 with aqueous HBF4 in benzene is thought to result in protonation at both Mo and the pendant phosphorus. Migration of the metal-hydride to the ring is followed by nucleophilic attack by OH−, resulting in C–P bond cleavage and formation of a phosphonium cation, [PH(OH)PhR]+. Further protonation of the metal centre and substitution of the remaining PPh2R ligand by P(OH)PhR gives the observed product.
![Suggested mechanism for the room temperature reaction of [Mo(Ph2P(CH2)nPPh2)(PPh2R)2] (21, R = Me, Ph; n = 1–3) with aqueous HBF4 in benzene.35](/image/article/2007/CS/b516248n/b516248n-f4.gif) | ||
| Fig. 4 Suggested mechanism for the room temperature reaction of [Mo(Ph2P(CH2)nPPh2)(PPh2R)2] (21, R = Me, Ph; n = 1–3) with aqueous HBF4 in benzene.35 | ||
4.3 OR derived from bidentate ligands
The observation of P–C bond cleavage in the [Pd(OAc)2]–PPh3 system has a long history, dating back to the early 1970s. Kikukawa and Matsuda noted that this combination would effect the phenylation of styrene and proposed that acetate could induce P–C/O exchange at Pd2+ centres via a nucleophilic 1,2-migration process.36 No intermediates were characterised, however, and more recent work by Amatore and Jutland has shown that the reaction of [Pd(OAc)2] with nPPh3 (n ≥ 2) results in disproportionation to give anionic Pd(0) species.4 It is possible that these are actually responsible for the P–C bond cleavage processes.An example where P–C/O exchange probably does proceed via a nucleophilic 1,2-migration process was described by Cotton in the reaction of the Ru2(II,III) tetraamidato species, 23, with triarylphosphines.37 In this case, the oxygen of the amidato ligand displaces an aryl group of PAr3 to give the Ru2(III,III) product, 24. The reaction is presumably initiated via Cl−/PAr3 substitution, but further mechanistic details could not be obtained for this complex process which, in addition to an oxidation of the Ru25+ core, involves multiple bond displacements and loss of two hydrogen atoms (25).

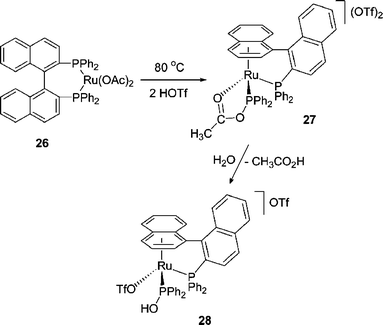
The best characterised example of P–C/O exchange induced by a bidentate ligand comes from the group of Pregosin through their studies of the acid-induced P–C bond cleavage reactions of Ru(P–P)(OAc)2 species (e.g., 26).38 In general, P–P represents the chiral chelating phosphines BINAP and MeO–BIPHEP) in these systems and similar results are obtained for both ligands. The discussion here and below is based on P–P = BINAP. With triflic acid, an initial product, 27, is formed that clearly demonstrates displacement by acetate of a naphthyl moiety from the BINAP ligand. After P–C bond cleavage, protonation of the naphthyl group must occur to produce the η6-arene group. Protonation also liberates acetic acid which subsequently produces water upon condensation to acetic anhydride. This water can then react with 27, effectively adding over the P–O bond to give 28. 28 can also be produced directly from 26 by performing the initial reaction with wet triflic acid.
4.4. OR derived from alcohols
Remarkably, the phosphinite complex 28 formed above undergoes further stereospecific reaction with alcohols to give double P–C/O exchange products, 29.38 This process is most rapid with smaller alcohols. Indeed, with tBuOH the expected Ph/OtBu exchange does not occur, but instead, reaction with trace water dominates to give dimeric products where both phenyl groups of the PPh2OH ligand have been replaced (30).
The conversion of 28 to 29 can also be reversed by protonation and a mechanism for these processes has been proposed (Fig. 5). From 28, initial solvolysis of the Ru–OTf bond forms a dicationic intermediate. Attack by ROH at phosphorus, with concomitant aryl transfer to ruthenium, then yields 29. The reverse process is driven by protonation.
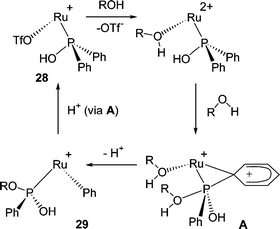 | ||
| Fig. 5 Proposed mechanism for the interconversion of 28 and 29. Non-participating ligands are omitted for clarity.38 | ||
With BINAP ligands bearing cyclohexyl or isopropyl substituents on phosphorus, analogues of both 28 and 29 are formed. Interestingly, however, in this case the reaction of the analogue of 28 with MeOH does not result in P–C/O exchange, but instead leads to a Ru–H species, presumably via β-H elimination.39
A related diastereoselective P–C/O exchange reaction had earlier been observed by Demerseman and co-workers upon heating diphenylphosphino-enolato complexes, such as 31, in methanol in the presence of potassium acetate.40

A mechanism involving solvolysis of the Ru–Cl bond in 31 was proposed so that upon nucleophilic attack of methanol at phosphorus transfer of one phenyl group to ruthenium may occur. The methanolic proton is subsequently removed by acetate. The nature of the base is important, however, as with K2CO3 no P–C/O exchange process occurs and Ru–hydride species are formed.
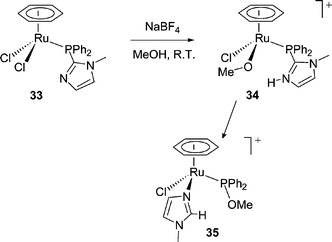
A similar reaction has been reported by Jalón and co-workers.41 Halide abstraction from 33 in methanol is postulated to give a methanol solvate within which internal proton transfer produces 34. Intramolecular nucleophilic attack by methoxide then occurs with displacement of the methylimidazolium moiety to the metal. The latter undergoes a tautomerisation to give the N-bound imidazole ligand in 35.
4.5 OR derived from alkoxide or aryloxide ligands
A well-characterised example implicating an alkoxide ligand in a P–C/OR exchange process was reported by van Leeuwen and co-workers. Reaction of Pt(Ph2PO)(Ph2POH)2H, 36, with o-(diphenylphosphino)benzaldehyde resulted in the formation of an oxaphosphole ligand in 38.42 This process is thought to involve substitution of one phosphinite ligand by the aldehyde and insertion into the Pt–H bond to give the metallacyclic alkoxide, 37. Nucleophilic attack of oxygen at phosphorus and a resultant 1,2-shift of a Ph group to Pt produces 38. It was noted at the time that ‘such P–C/Pt–O metathesis may have been somewhat overlooked as a phosphine decomposition mode’. A related process has also been noted at Rh.43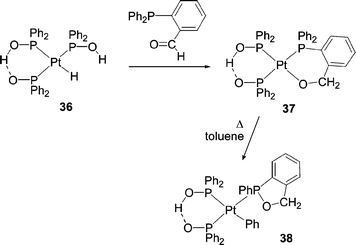
Facile aryl/aryloxide exchange has been proposed recently in the reactions of [PdCl2(PPh3)2] with NaOAr in THF.44 NMR studies provide evidence for the initial formation of [PdCl(OAr)(PPh3)2] at 0 °C, but this rapidly gives way to [PdCl(Ph)(PPh3)2] upon warming. In this case, the P–C/O exchange process is postulated to occur via a phosphonium salt, [PPh3(OAr)]+[PdClPPh3]−. Interestingly, many analogous [PdCl(OAr)L2] species are known where L2 features alkylphosphines or bidentate phosphines. The PPh3 system therefore appears particularly vulnerable to OAr-induced substituent replacement.
In the light of this, the observation of P–C/O exchange in [Ru(OC6H4–p-Me)2(PMe3)4], 39, is all the more remarkable as it involves cleavage of a simple, unstrained P–C(sp3) bond.45 A mechanism based on initial PMe3 loss, followed by the oxidative addition of one P–Me bond to Ru, has been proposed. P–O bond forming reductive elimination would form a PMe2(OC6H4–p-Me) ligand which would undergo orthometallation to give the final product, 40.

5. P–C/F exchange reactions
Until recently P–C/F exchange reactions were rare, but in the last 5 years a number of well-defined examples have been published. Moreover, both experimental and computational mechanistic studies are beginning to shed light on the detailed pathways involved in these intriguing reactions.Some known P–C/F exchange processes bear close relation to certain P–C/OR exchange reactions discussed above. For example, when [Mo(Ph2P(CH2)nPPh2)(PPh2R)2] species (21, R = Me, Ph; n = 1–3) are protonated with HBF4·Et2O in the absence of water the formation of fluorophosphine complexes analogous to 22 is seen (see Fig. 4). A similar mechanism to that proposed for the analogous phosphinite complexes was suggested with, in this case, F− (originating from BF4−) acting as the nucleophile.35
Another case where the ‘inert’ BF4− counterion acts as a F− source was seen by Pregosin and Geldbach upon protonation of Ru(P–P)(OAc)2 species (P–P = BINAP or MeO–BIPHEP).38 Indeed, this was the original observation that eventually led to the characterisation of the wide range of P–C/OR exchange processes discussed in Sections 4.4 and 4.5. In this instance, low temperature protonation of both acetate ligands occurs, with one being replaced by fluoride to give 41. Warming 41 then induces a P–C/F exchange process where the naphthyl group is again protonated to generate an η6-arene ligand. An important feature of the intermediate 41 is the additional η2-interaction from one of the naphthyl moieties with the Ru centre and this precoordination is thought to promote the P–C cleavage process.

In related work, reaction of the cationic derivatives [RuCl(η-arene)(P–P)]+ (arene = benzene, p-cymene) with [NBu4][F2SiPh3] as a fluoride source leads to the isolation of direct fluoro/naphthyl exchange products exemplified by 43.46

Several P–C/F exchange reactions are known for which no parallel P–C/OR exchange has been seen. Milstein and co-workers found that heating [IrMe(PEt3)3], 44, in hexafluorobenzene led to C–F activation.47 In addition, however, this was coupled not only to P–C/F exchange, but also to the elimination of methane and ethene to give the eventual product, trans-[Ir(C6F5)(PEt3)2(PEt2F)], 45.

At the time, a mechanism based on the initial metallation of one PEt3 ligand followed by electron transfer to C6F6 was proposed, based primarily on the lack of reaction with lower fluorinated arenes. However, more recent density functional calculations on a model trans-Ir(PH3)2(PH2Et)Me system, 44′, suggest that C–F bond activation may occur in a concerted fashion over an Ir–P bond to generate a metallophosphorane intermediate, 46.48 Such a step would be closely related to SNAr processes and is thus also consistent with the greater reactivity of C6F6. In this case, a phosphine ligand performs as a Lewis acid and traps out the displaced F− anion.49

In the course of their extensive studies on the C–F bond activation reactions of fluorinated heterocycles, Perutz, Braun and co-workers found that the reaction of pentafluoropyridine with [Pt(PR3)2] (R = Cy, iPr) in THF leads to the unusual P–C/F exchange product, 47.50

Possible mechanisms were discussed for this process, based on the rearrangement of an initial C–F bond activation product, trans-[Pt(4-C5NF4)F(PR3)2]. Importantly, P–C/F exchange is solvent dependent and does not occur in hexane. This was thought to indicate the presence of charged intermediates, such as the ion pair [PFR3]+[Pt(4-C5NF4)(PR3)]− or Pt–phosphido species such as [Pt(4-C5NF4)(PR2)(R)(PR3)]+F−. Analogous P–C/F exchange chemistry was also observed subsequently by Grushin et al. in the reaction of C6F6 with [Pt(PR3)2] species.49
Perhaps the best characterised P–C/F exchange process has been reported recently by Grushin and Marshall.51 They found that heating [RhF(PPh3)3], 48, in chlorobenzene led to the fluorophosphine complex trans-[RhCl(PFPh2)(PPh3)2], 50, and biphenyl. Further mechanistic studies defined a F/Ph exchange species, cis-[RhPh(PFPh2)(PPh3)2], 49, as an intermediate.49 Kinetic studies showed that 49 is formed via an intramolecular P–Ph/F exchange process with ΔH‡ = 22.0 ± 1.2 kcal mol−1 and ΔS‡ = −10.0 ± 3.7 eu. In addition, the 48 to 49 interconversion is not influenced by added phosphine. Once formed, 49 is sufficiently electron-rich to activate the C–Cl bond of chlorobenzene with subsequent elimination of biphenyl giving 50.

Density functional calculations on a cis-[RhF(PH3)2(PH2Ph)] model system, 48′, considered two processes for the P–C/F exchange (Fig. 6). Pathway 1 is based on Mechanism A of Fig. 1 and involves Ph transfer from P to Rh followed by P–F bond forming reductive elimination. Pathway 2 assesses Mechanism E, where F acts as an intramolecular nucleophile to give a metallophosphorane intermediate from which Ph migration to Rh completes P–C/F exchange.
![Computed mechanisms for P–C/F exchange in cis-[RhF(PH3)2(PH2Ph)], 48′.](/image/article/2007/CS/b516248n/b516248n-f6.gif) | ||
| Fig. 6 Computed mechanisms for P–C/F exchange in cis-[RhF(PH3)2(PH2Ph)], 48′. | ||
With these small model systems both pathways were computed to have similar activation barriers of around 31 kcal mol−1, although the metallophosphorane pathway was favoured on the basis of differential solvation effects and the fact that, experimentally, [IrF(PPh3)3] (for which an Ir(III)–phosphido intermediate might be expected to be more accessible than for its Rh(III) analogue) did not exhibit any P–C/F exchange chemistry. Calculations on the full Rh(PPh3)3F system have subsequently shown a clear preference for the metallophosphorane pathway.52
Outlook
A wide range of P–C/X exchange processes has now been identified. Of these, P–aryl/aryl exchange appears to be the most common and this process seems to be systematically present as a side reaction in many metal-mediated cross-coupling reactions. The nature of the phosphine substituent is clearly important and P–R/X exchange is far more common for triaryl- than trialkylphosphines. Mechanistic studies indicate that P–Ar/Ar′ exchange is promoted when both Ar and Ar′ are electron-rich groups. However, controlling P–Ar/Ar′ exchange will not always be compatible with retaining high catalytic activity.For other X groups, NR2, OR and F, the observation of P–R/X exchange has often been serendipitous. However, more well-defined examples of these processes are now appearing and patterns are beginning to emerge. The relative paucity of examples of P–R/X exchange when X = NR2 and, until recently, X = F, may simply reflect the difficulty in the synthesis of complexes that combine both phosphine and hard donor ligands. Indeed, such exchange processes, in conjunction with related disproportionation processes,4–6 may be reasons why such species are so difficult to prepare. A better understanding of P–C/X exchange processes therefore promises an insight into how such ‘mismatched’ low-valent transition metal–phosphine complexes may be stabilised in the presence of hard NR2, OR and F ligands.
References
- P. E. Garrou, Chem. Rev., 1985, 85, 171 CrossRef CAS and references therein.
- A. W. Parkins, Coord. Chem. Rev., 2006, 250, 449 CrossRef CAS.
- P. W. N. M. van Leeuwen, Appl. Catal., A, 2001, 212, 61 CrossRef CAS.
- C. Amatore and A. Jutland, Acc. Chem. Res., 2000, 33, 314 CrossRef CAS and references therein.
- V. V. Grushin and H. Alper, Organometallics, 1993, 12, 1890 CrossRef CAS.
- M. R. Mason and J. G. Verkade, Organometallics, 1992, 11, 2212 CrossRef CAS.
- R. A. Dubois and P. E. Garrou, Organometallics, 1986, 5, 466 CrossRef CAS.
- A. G. Abatjoglou and D. R. Bryant, Organometallics, 1984, 3, 932 CrossRef CAS.
- A. B. Goel, Inorg. Chim. Acta, 1984, 86, L77 CrossRef CAS.
- K.-C. Kong and C.-H. Cheng, J. Am. Chem. Soc., 1991, 113, 6313 CrossRef CAS.
- B. E. Segelstein, T. W. Butler and B. L. Chenard, J. Org. Chem., 1995, 60, 12 CrossRef CAS.
- M. Sakamoto, I. Shimizu and A. Yamamoto, Chem. Lett., 1995, 24, 1101 CrossRef.
- G. de la Torre, A. Gouloumis, P. Vázquez and T. Torres, Angew. Chem., Int. Ed., 2001, 40, 2895 CrossRef CAS.
- F. E. Goodson, T. I. Wallow and B. M. Novak, J. Am. Chem. Soc., 1997, 119, 12441 CrossRef CAS and references therein.
- V. V. Grushin, Organometallics, 2000, 19, 1888 CrossRef CAS and references therein.
- W. A. Herrmann, C. Brossmer, K. Öfele, M. Beller and H. Fischer, J. Mol. Catal. A: Chem., 1995, 103, 133 CrossRef CAS.
- D. F. O'Keefe, M. C. Dannock and S. M. Marcuccio, Tetrahedron Lett., 1992, 33, 6679 CrossRef CAS.
- K. R. Buszek and Y.-M. Jeong, Tetrahedron Lett., 1995, 36, 5677 CAS.
- D. Barañano and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 2937 CrossRef CAS.
- A. G. Abatjoglou, E. Billig and D. R. Bryant, Organometallics, 1984, 3, 923 CrossRef CAS.
- J. V. Ortiz, Z. Havlas and R. Hoffmann, Helv. Chim. Acta, 1984, 67, 1 CAS.
- M. L. H. Green, M. J. Smith, H. Felkin and G. Swierczewski, J. Chem. Soc. D, 1971, 158 Search PubMed.
- R. Mohtachemi, G. Kannert, H. Schumann, S. Chocron and M. Michman, J. Organomet. Chem., 1986, 310, 107 CrossRef CAS.
- D. K. Morita, J. K. Stille and J. R. Norton, J. Am. Chem. Soc., 1995, 117, 8576 CrossRef CAS.
- P. Vierling and J. G. Riess, Organometallics, 1986, 5, 2543 CrossRef CAS and references therein.
- S. Al-Jibori, W. S. MacDonald and B. L. Shaw, J. Chem. Soc., Chem. Commun., 1982, 287 RSC.
- S. Al-Jibori, M. Hall, A. T. Hutton and B. L. Shaw, J. Chem. Soc., Chem. Commun., 1982, 1069 RSC.
- S.-E. Bouaoud, P. Braunstein, D. Grandjean, D. Matt and D. Nobel, Inorg. Chem., 1986, 25, 3765 CrossRef CAS.
- M. Cowie, I. R. McKeer, S. J. Loeb and M. D. Gauthier, Organometallics, 1986, 5, 860 CrossRef CAS.
- D. V. Naik, G. J. Palenik, S. Jacobson and A. J. Carty, J. Am. Chem. Soc., 1974, 96, 2286 CrossRef CAS.
- The nature of the O-donor is also a factor as P–C/O exchange has been reported upon reaction of [Pt(PPh3)3] with nitrile oxides to give a Pt–diphenylphosphine oxide complex. W. Beck, M. Keubler, E. Leidl, U. Nadel, M. Schaal, S. Cenini, P. Del Buttero, E. Licandro, S. Maiorana and A. C. Villa, J. Chem. Soc., Chem. Commun., 1981, 446 Search PubMed.
- N. W. Alcock, P. Bergamini, T. J. Kemp and P. G. Pringle, J. Chem. Soc., Chem. Commun., 1987, 235 RSC.
- I. J. B. Lin, J. S. Lai and C. W. Liu, Organometallics, 1990, 9, 530 CrossRef CAS.
- V. Cadierno, J. Díez, J. Garcia-Álvarez and J. Gimeno, Organometallics, 2004, 23, 3425 CrossRef CAS.
- R. H. Morris, J. F. Sawyer, C. T. Schweitzer and A. Sella, Organometallics, 1989, 8, 2099 CrossRef CAS.
- K. Kikukawa and T. Matsuda, J. Organomet. Chem., 1982, 235, 243 CrossRef CAS and references therein.
- A. K. Chakravarty and F. A. Cotton, Inorg. Chem., 1985, 24, 3584 CrossRef CAS and references therein.
- T. J. Geldbach and P. S. Pregosin, Eur. J. Inorg. Chem., 2002, 1907 CrossRef CAS and references therein.
- T. J. Geldbach, P. S. Pregosin and A. Albinati, Organometallics, 2003, 22, 1443 CrossRef CAS.
- P. Crochet, B. Demerseman, C. Rocaboy and D. Schleyer, Organometallics, 1996, 15, 3048 CrossRef CAS.
- A. Caballero, F. A. Jalón, B. R. Manzano, G. Espine, M. Pérez-Manrique, F. J. Poblete and M. Maestro, Organometallics, 2004, 23, 5694 CrossRef CAS.
- P. W. N. M. van Leeuwen, C. F. Roobeek and A. G. Orpen, Organometallics, 1990, 9, 2179 CrossRef CAS.
- D. Selent, W. Baumann, R. Kempe, A. Spannenberg, D. Röttger, K.-D. Wiese and A. Börner, Organometallics, 2003, 22, 4265 CrossRef CAS.
- H. Yasuda, N. Maki, J.-C. Choi, M. Abla and T. Sakakura, J. Organomet. Chem., 2006, 691, 1307 CrossRef CAS.
- J. F. Hartwig, R. G. Bergman and R. A. Andersen, J. Organomet. Chem., 1990, 394, 417 CrossRef CAS.
- C. J. den Reijer, P. Dotta, P. S. Pregosin and A. Albinati, Can. J. Chem., 2001, 79, 693 CrossRef CAS.
- O. Blum, F. Frolow and D. Milstein, J. Chem. Soc., Chem. Commun., 1991, 258 RSC.
- S. Erhardt and S. A. Macgregor, unpublished results.
- S. A. Macgregor, D. C. Roe, W. J. Marshall, K. M. Bloch, V. I. Bakhmutov and V. V. Grushin, J. Am. Chem. Soc., 2005, 127, 15304 CrossRef CAS.
- N. A. Jasim, R. N. Perutz, A. C. Whitwood, T. Braun, J. Izundu, B. Neumann, S. Rothfeld and H.-G. Stammler, Organometallics, 2004, 23, 6140 CrossRef CAS and references therein.
- V. V. Grushin and W. J. Marshall, J. Am. Chem. Soc., 2004, 126, 3068 CrossRef CAS.
- S. A. Macgregor and T. Wondimagegn, unpublished results.
| This journal is © The Royal Society of Chemistry 2007 |