Micropatterned supported membranes as tools for quantitative studies of the immunological synapse
Kaspar
Mossman
abc and
Jay
Groves
*abc
aBiophysics Graduate Group, University of California, Berkeley, California, USA
bDepartment of Chemistry, University of California, Berkeley, California, USA
cPhysical Biosciences and Material Sciences Divisions, Lawrence Berkeley National Laboratory, Berkeley, California, USA. E-mail: kdmossman@gmail.com; jtgroves@lbl.gov; Fax: +1-510-643-6232; Tel: +1-510-643-0186
First published on 14th September 2006
Abstract
In living cells, membrane receptors transduce ligand binding into signals that initiate proliferation, specialization, and secretion of signaling molecules. Spatial organization of such receptors regulates signaling in several key immune cell interactions. In the most extensively studied of these, a T cell recognizes membrane-bound antigen presented by another cell, and forms a complex junction called the “immunological synapse” (IS). The importance of spatial organization at the IS and the quantification of its effect on signaling remain controversial topics. Researchers have successfully investigated the IS using lipid bilayers supported on solid substrates as model antigen-presenting membranes. Recent technical developments have enabled micron- and nanometre-scale patterning of supported lipid bilayers (SLBs) and their application to immune cell studies with provocative results, including spatial mutation of the IS. In this tutorial review, we introduce the IS; discuss SLB techniques, including micropatterning; and discuss various methods used to perturb and quantify the IS.
 Kaspar Mossman | Kaspar Mossman studied physics at Mount Allison University and the University of British Columbia before designing superconducting filters as a radio-frequency engineer in Santa Barbara, California. After developing an interest in biological systems, he began a PhD in biophysics at the University of California, Berkeley, in 2001. He graduated in May 2006 and is now working in science publishing. |
 Jay Groves | Jay Groves received a BSc in Physics and Chemistry from Tufts University and studied with Steve Boxer at Stanford University for his PhD, which he received in 1998. After a year as a Visiting Scholar at the Academia Sinica in Taipei, he returned to California for an appointment as a Division Director’s Fellow at Lawrence Berkeley National Laboratory. In 2001 he was appointed Assistant Professor of Chemistry at the University of California, Berkeley. His research interests focus on interfacial membrane systems, both living and synthetic, in which collective, large-scale properties influence the molecular chemistry. |
Acronyms used: IS, immunological synapse; SLB, supported lipid bilayer; TIRF, total internal reflection microscopy; GPI, glycosylphosphatidylinositol; APC, antigen-presenting cell; MHC, major histocompatibility complex; pMHC, peptide-MHC; SMAC, supramolecular activation cluster; cSMAC, central SMAC; pSMAC, peripheral SMAC; LFA, lymphocyte function-associated antigen; TCR, T cell receptor; GFP, green fluorescent protein; ICAM, intercellular adhesion molecule; ITAM, immunotyrosine activation motif; RBL, rat basophilic leukemia.
General introduction
Spatial organization of membrane receptors for the regulation of intracellular signaling is common to many different biological systems, particularly in the immune system. Clustering of receptors into multimeric and supramolecular complexes drives chemotactic responses in bacteria,1 degranulation of mast cells,2 and information transfer between lymphocytes.3,4 The evolution of molecular patterns as cells interact with soluble and membrane-bound ligands can be observed with submicron resolution using fluorescence microscopy. For imaging, cells may be genetically modified to produce fluorescent receptors; or fluorophores may be covalently linked to ligands or antibodies, which bind receptors. While in vivo observations may provide the most natural and complete picture of cellular behaviour, in vitro experimental systems allow researchers to control the chemical and topological environment, and to perturb cells in a controlled manner along a variety of experimental dimensions.In particular, planar artificial biointerfaces of lipids, proteins, and polymers may be fabricated using techniques developed by the semiconductor industry.5,6 A planar geometry permits optical imaging at the highest possible resolution, by epifluorescence or total internal reflection microscopy (TIRF)—which makes possible imaging of the dynamics of single molecules—in contrast to confocal imaging of randomly oriented cell–cell interfaces, which is much slower and limited to a resolution of ∼1 µm in the z-dimension. Surfaces patterned from immobile material such as extracellular matrix7 or elastomer8 may be used to quantify cytoskeletal forces or investigate the relationship between cell attachment and survival. However, supported bilayer membranes of phospholipids and GPI- or lipid-linked proteins provide a closer mimic of cell plasma membranes. Most importantly, their molecular constituents are able to diffuse freely, which allows adherent cells to bind and spatially reorganize ligands. Combined with lithographic patterning techniques, supported membranes are powerful tools which researchers are using to explore the connection between spatiotemporal organization and signaling.
The immunological synapse
T lymphocytes comprise a key division of the adaptive immune response. Each T cell expresses many copies of one, and only one, antigen receptor in its plasma membrane, which bind with great specificity to antigen fragments held in the groove of major histocompatibility protein (MHC) on the surface of antigen-presenting cells (APCs). Upon recognizing specific antigen, naive (previously unexposed) T cells can become activated, and amplify the immune response by proliferation and differentiation to specialized cell types. Armed “effector” T cells that recognize antigen carry out functions such as destruction of viral-infected APCs (in the case of CD8+ cytotoxic T cells) or cytokine release to stimulate B cell antibody production or recruit macrophages (in the case of CD4+ helper T cells).The binding of T cell receptors (TCRs) to peptide-MHC (pMHC) triggers nucleation of clusters of signaling and cytoskeletal molecules, which assemble into a central supramolecular activation cluster (cSMAC), surrounded by a broad ring of integrins such as LFA-1, the peripheral SMAC (pSMAC) (see Fig. 1). This junction is called an “immunological synapse” (IS) because of its similarity to a neural synapse. While the neural synapse is a permanent structure, the immunological synapse exists only for minutes or hours. Nevertheless, they share a common theme: a tight cell–cell interface across which information is transferred. In the neural synapse, transmission takes the form of neurotransmitter exocytosis. There is a direct parallel in the immunological synapse, as cytokines or vesicles containing cytolytic substances may be transferred from T cell to the APC, although the primary information transfer occurs through TCR-pMHC binding. Since the first observations of the helper T cell IS, similar structures have been found in many instances of communication in the immune system. Immature T cells (thymocytes) form multifocal, dynamic synapses;9 cytotoxic T cells form synapses with target cells;10 “natural killer” cells11 and B cells12 also transfer information via synapse. The CD4+ helper T cell synapse is the best-characterized, experimentally, and the results discussed in this review will focus on that system.
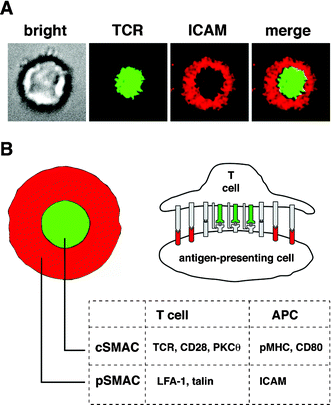 | ||
| Fig. 1 The immunological synapse. (A) Brightfield and fluorescence images of a T cell forming a synapse with a supported membrane containing GPI-linked pMHC and ICAM. (B) Schematic and cross-section of the mature synapse showing segregation of signaling molecules in the cSMAC and adhesion molecules in the pSMAC. | ||
Monks et al. first observed the striking pattern of the mature IS by confocal microscopy of 3D interactions between T cells and APCs.3 Using antibodies, they labeled the signaling marker protein kinase C-θ (in the cSMAC) and the cytoskeletal linker talin (in the pSMAC) on the cytosolic side of the T cell. Their seminal paper highlighted both the variety of molecular involvement at the membrane and in the cytosol as well as an apparent importance for the cSMAC in TCR-specific signaling. Grakoui et al., with a simplified system—a planar supported lipid bilayer incorporating aliphatically coupled proteins, that substituted for the APC membrane4—showed that the IS is a dynamic assembly which evolves from a diffuse distribution of receptors and ligands to a highly organized, largely static structure. As the synapse evolves, certain important components migrate either inward or outward, suggesting that spatiotemporal organization may regulate signaling.
Several other papers have presented results that underline the dynamic nature of the synapse. Krummel et al. transfected CD4+ T cells with GFP-fused CD4 and CD3ζ (a component of the TCR complex).13 They observed that while CD3ζ clustered in a stable cSMAC, CD4, which initially co-localized with CD3ζ, dispersed to the periphery after 2 minutes. They suggest that CD4 is required for initial TCR triggering, but not for sustained signaling. In this same paper, they observed that the cSMAC formed by accumulation of small, dynamic TCR clusters. Recent results suggest that a continual flow of these microclusters may sustain signaling over the hours necessary to induce full activation.14 Lee et al., imaging naive CD4+ T cells interacting with APCs, found that TCR-mediated tyrosine kinase signaling began before formation of the mature IS, and was concentrated in the periphery.15 They suggest that either the cSMAC is required for TCR down-regulation and endocytosis or that it provides an environment for activation of other receptors. Freiberg et al., obtaining 3D fluorescence images of CD4+ blasts forming synapses with B cells, found that CD45 was recruited to the cSMAC by the 3 minute point, and then excluded to a previously unidentified distal SMAC, or dSMAC, in the extreme periphery of the synapse.16 pY signaling in the cSMAC resumed after CD45 exclusion. Freiberg et al. claim that CD45 serves to synchronize signaling in the cSMAC, because kinases and ligands may arrive in the cSMAC in different states of phosphorylation; this might prevent spurious signaling from kinases binding TCRs not encountering cognate pMHC.
Supported lipid bilayers (SLBs)
Phospholipids comprise a major component of cell membranes. Bilayers reconstituted from lipid components are ideal experimental systems either for studies of fundamental membrane properties such as phase behavior or fluctuations, or for investigation of cellular adhesion or membrane-proximal signaling. Membranes supported on solid substrates are convenient as cell mimics, if spherical silica beads are used, or for imaging at high resolution or with advanced techniques such as total internal reflection microscopy, if planar substrates are used. The key feature of the supported membrane is fluidity. Individual lipids retain high diffusive mobility. Fluid membranes may be supported on thin, hydrated polymer cushions;17 the advantage of this system is that transmembrane proteins may be incorporated, and retain mobility. Transmembrane proteins in membranes supported only by a water layer are immobile because the cytosolic domain adheres to the hydrophilic substrate. Protein components linked to lipid or GPI anchors, however, retain mobility in such membranes (see Fig. 2).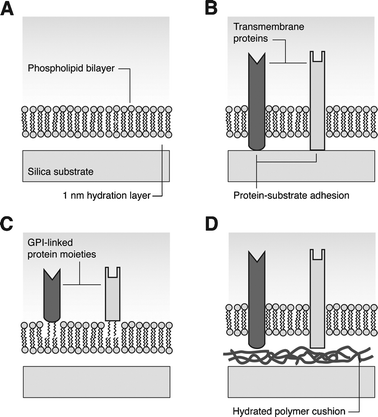 | ||
| Fig. 2 Solid-supported lipid bilayers. (A) Amphiphilic phospholipids form a bilayer separated from a hydrophilic substrate by a thin hydration layer. (B) Transmembrane proteins embedded in the bilayer adhere to the substrate and are immobilized. (C) Extracellular protein moieties coupled to GPI, incorporated in the bilayer, retain fluid properties. (D) If the bilayer is supported on a hydrated polymer cushion, transmembrane proteins remain fluid. | ||
There are several methods by which SLBs may be formed. Alkylated substrates, or substrates such as glass, quartz, mica, or single-crystal silicon made hydrophilic by an etching step, may be lowered vertically through a phospholipid monolayer in a Langmuir trough (and in the case of the hydrophilic substrates, raised up and re-lowered horizontally).18 For membranes which are to incorporate substantial quantities of protein, membrane formation by vesicle fusion is more appropriate, to avoid protein oxidation or denaturation. Transmembrane or GPI-linked proteins are solubilized and mixed with detergent-solubilized phospholipids, and the detergent removed by dialysis. To form a membrane, the vesicles are introduced to a hydrophilic substrate; they adsorb, rupture, and fuse into a continuous membrane which is trapped close to the substrate in an energy minimum produced by a balance of attractive van der Waals forces with repulsive electrostatic and hydration forces.5 The membrane is separated from the substrate by a thin (∼10 nm) layer of water, which preserves lipid fluidity.19
Planar SLBs in immune cell studies
Planar SLBs have long been used as analogs of cell membranes in studies of adhesion and communication of immune cells.20 Early work demonstrated that cytotoxic T cells could be activated by immobile pMHC incorporated in a bilayer;21 fluorescence resonance energy transfer in binding between T cells and pMHC in a SLB was used in a key experiment to show that TCR could stabilize peptide interaction with MHC.22 Quantitative studies of adhesion forces between CD2 and CD58 were made with Jurkat T cells and SLBs.23 Soon afterward, it was discovered that CD58 existed in vivo in two forms, both transmembrane and associated to the membrane via a GPI linkage. This important finding spurred genetic work to develop hybrid macromolecules consisting of the extracellular moieties of MHC and ICAM-1 linked to GPI, and thus capable of being incorporated into a bilayer and maintaining molecular fluidity, unlike transmembrane versions of the same proteins. Grakoui et al. were then able to perform their canonical experiments with immunological synapses formed between T cells and SLBs containing GPI-linked pMHC and ICAM-1.4On a parallel experimental path, researchers recently demonstrated a neural-type synapse formed between HEK cells expressing neurexin and a SLB incorporating GPI-linked neuroligin.24
An alternative to incorporation of GPI-linked or transmembrane proteins is modification of a phospholipid headgroup by covalent linkage of a hapten such as dinitrophenol, which in conjunction with the phospholipid head creates an epitope that can be recognized by an antibody.25 This provides an experimental platform for investigation of Fc receptor-triggered responses of mast cells, which are important in inducing inflammation. The multimeric FcεRI receptor has a high affinity for extracellular IgE; crosslinking by multivalent antigen initiates phosphorylation of FcεRI immunotyrosine activation motifs (ITAMs) similar to those in the CD3ζ chains of the TCR, by the Src-family kinase Lyn. The similarity of the initial stages of TCR and FcεRI signaling at the membrane has led some researchers to use the FcεRI system as a simpler model of the immunological synapse, with special focus on the spatiotemporal organization of lipid rafts (liquid-ordered membrane domains enriched in cholesterol and sphingomyelin) as a regulator of receptor signaling.26
Quantification of IS phenomena
With a system that resembles an analog computer as much as the IS does, there is strong interest in quantifying input, output, and the intervening “calculations” carried out by intracellular networks.Inputs to the T cell consist of peptide-MHC density and composition; costimulatory signals; size of TCR clusters; and spatial organization of membrane molecules. Quantification of inputs is usually achieved through fluorescence microscopy. One great advantage of the hybrid T cell-SLB system is that all the synaptic interactions take place in a well-defined optical plane; further, confocal microscopy can be used to eliminate out-of-plane fluorescence. However, even at highest resolution, imaging of molecular clusters is diffraction-limited. Also, photobleaching is a problem. TCR microclusters are submicron in diameter, and labeling fluorophores bleach quickly if illuminated by a mercury lamp. In consequence, the literature contains far more bulk measurements of molecular density, or observations of large-scale exclusion or colocalization, than observations of single-molecule or cluster dynamics.
Although it is possible to control the density of GPI-linked ligands in a SLB, its fluidity enables synapsing T cells to accumulate concentrations of pMHC above background. Grakoui et al. used fluorescence intensity data to determine the density of MHC in the synapse as well as the time course of its accumulation.4 They found a strong linear correlation between the half-time of TCR-pMHC binding and total MHC accumulation. They also noted that a minimum density of 60 pMHC µm−2 in the cSMAC was necessary to induce proliferation.
Several groups have investigated the dynamics of TCR motion in the IS, both as single molecules and clusters. Moss et al. developed a method for tracking TCRs in three dimensions as T cells made contact with APCs, and found that the range of TCR velocities significantly exceeded what would be expected for diffusion.27 Yokosuka et al. performed a comprehensive study of microcluster accumulation in the cSMAC, tracking the distinctive radial paths of CD3ζ, ZAP70, and the cytoskeletal adaptor SLP-76 and correlating cluster motion with calcium flux.14 In earlier work, Krummel et al. had quantified the number of CD4 clusters in the cSMAC to demonstrate the delay in exclusion to the periphery.13 An early and convincing argument for the cytoskeleton as the source of organization in the IS was made by Wulfing et al., who attached silica beads to lipids and transmembrane proteins in the T cell membrane, and measured bead velocity as directed motion began upon antigen recognition.
Intermediate stages in activation, such as kinase activity that results in phosphorylation of key tyrosine residues and hence activation of signaling cascades, can be observed using fluorescent chimeric proteins or fix-and-stain antibody methods. The key results obtained in this manner, focused on the time dependence of bulk molecular distributions, have been described earlier in this review.13,15,16 A strong theme that emerges from these results is the IS as a dynamic three-dimensional pattern—two spatial dimensions, plus time. Downstream of the synapse itself, cytosolic calcium levels, which regulate many cellular processes through calcium-dependent intermediates such as calcineurin, are of particular interest. Calcium clamping experiments have correlated both frequency and mean level of Ca oscillations in T cells with altered cytokine output.28 A thorough mathematical treatment of the statistics of time-dependent Ca signaling in T cells revealed that higher-frequency oscillations are associated with interferon-γ production.29
Interpretation of time-dependent data must take into account the variation in behavior over a population of cells, which may be differentiated in type or at different stages in the cell cycle. A population of cells (typically, several million) will not be synchronized, and therefore data collected live must be calibrated from the instant at which the T cells make contact with APC or SLB. This is often taken to be coincident with the characteristic large spike in intracellular calcium when a T cell encounters cognate antigen. In fix-and-stain experiments, in which cellular processes are immobilized with formaldehyde and labeled with fluorescent antibodies, it is difficult to interpret data taken in the first minute after cells are introduced to the experimental volume. Some cells may be frozen seconds after contact, while others may be captured well into synapse formation. To avoid this uncertainty, there is a trend toward the use of live experiments using fluorescent fusion proteins, such as GFP-CD4,13 which are introduced into cells by DNA transfection. Individual molecules or cells can be observed to the limit that equipment and photobleaching allow. However, biological understanding emerges only in the statistical behavior, embodied as means and standard deviations, of a relatively large number of cells in repeated experiments.
If all cells are presented with the same membrane stimulus (generally not possible with patterns fabricated by electron-beam lithography, because of the limited area), T cell output can be assessed by the frequency of cell division, as well as the quantity of growth factors and cytokines secreted. Supernatant can be sampled and secreted factors identified with an enzyme-linked immunosorbent assay. Proliferation can be determined by measuring incorporation of radioactive thymidine4 or by loading cells with the fluorescent dye carboxyfluorescein succinimidyl ester (CFSE), the intensity of which is halved upon successive cell divisions.15
Although a T cell is an extremely complex system, the IS itself is a more tractable subsystem in which membrane properties and binding kinetics are coupled with molecular transport, driven by passive as well as active (motor protein) mechanisms. Computational chemists consider the IS a paradigm of spatial regulation of signaling and have developed quantitative models to explore both synapse formation and signal processing. The advantage of a quantitative model is that it allows theorists to understand the IS and its many molecular interactions on a more sophisticated level than the usual biological model, which consists of a diagram of proteins connected by lines indicating that they activate and deactivate each other.
Qi et al. developed a mathematical description of the IS30 in which an initial homogenous receptor distribution at the interface between a parabolic membrane (the T cell) and a planar membrane (the APC) is capable, as it moves toward equilibrium, of evolving into spatiotemporal patterns which resemble an IS. Subsequently, S.-J. Lee et al. used a similar model to demonstrate that the multifocal, transient synapses observed in thymocytes could be the result of lower TCR expression.31 The most comprehensive model so far has been that of K. H. Lee et al., who combined a computational model of receptor distribution and signaling networks with genetic manipulation and live cell experiments.32 They found that the large accumulation of pMHC in the cSMAC served both to sustain weak signaling (due to low TCR-pMHC affinity, or low TCR expression) and to shut down strong signaling (by endocytosis of TCRs once activated): clustering of pMHC in a cSMAC enhances the probability that a TCR will rebind pMHC after dissociation.
Micropatterning SLBs for cell studies
Planar SLBs may be applied in studies of fundamental cellular phenomena or as biosensors.33 For both purposes, it is useful to control their spatial properties, especially at subcellular length scales. There exist many methods by which SLBs may be patterned, which usually involve photolithography with its potential for high throughput (e.g. selective UV exposure to photochemically degrade lipids; microcontact inking and stamping). However, only two methods have successfully been applied to immune cell studies: polymer liftoff and diffusion barriers (see Fig. 3).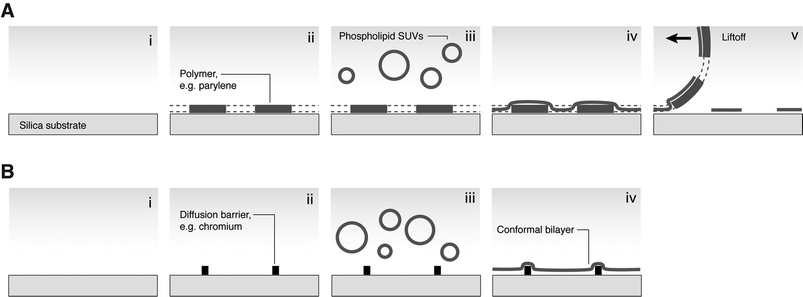 | ||
| Fig. 3 Methods of micropatterning SLBs for cell studies. (A) Polymer liftoff. (i) Hydrophilic substrate; (ii) photolithographically patterned polymer layer; (iii) SLB deposition from vesicles; (iv) conformal bilayer; (v) polymer is peeled off, leaving patterned membrane. (B) Diffusion barriers. (i) Hydrophilic substrate; (ii) barriers patterned by photo- or electron-beam lithography; (iii) SLB deposition; (iv) conformal bilayer. Lipids on barriers are immobile and lipids in corrals are unable to diffuse across barriers. | ||
Arrays of SLB regions can be produced by liftoff of a polymer such as parylene. The polymer is patterned by photolithography on a SiO2 substrate; then the lipid membrane is deposited; then the polymer is peeled off, to leave fluid lipid islands which hold their shape. The resolution of this technique has improved to the micron level.34 The diffusion barrier approach is amenable to both photolithography and electron-beam lithography.35 SLBs are deposited on a hydrophilic substrate on which lithographically defined patterns have been fabricated. By this method, micron-scale arrays of membrane corrals may be defined, which are spatially addressable. Barriers may be fabricated from protein, photoresist, or many inorganic materials, especially metals.36 Electrostatic or chemical properties, and not topography, are responsible for the barrier action, since fluid membranes will spread over and conform to SiO2 barriers, while barriers of similar dimensions fabricated from metal will block diffusion. For the research described here, the most significant advantage of barrier-partitioned SLBs is that the barriers may be defined by electron-beam lithography, and therefore made extremely small (∼100 nm), so that their width is negligible compared with the diameter of a large cell such as a T lymphocyte.
Methods of perturbing the IS
Experimentalists have several options if they wish to perturb the IS: agents to block signaling (such as antibodies or drugs), cytoskeletal interactions (such as cytochalasin D or latrunculin), or genetic manipulations. With hybrid cell/membrane systems, there is the possibility of constraining the ligands/lipids present in the biointerface, and directly influencing spatial organization.All these methods—which may of course be used in combination—have advantages and drawbacks. Genetically transformed T cells may be observed in vivo, operating in a near-natural chemical and biological environment (e.g., with in vivo cytokine levels, and ligand stimulation available from stromal cells). However, many, if not all, proteins involved in the IS have multiple interactions and functions. For example, Lck binds to CD4, phosphorylates both CD3ζ and ZAP70, has an inositol trisphosphate (IP3) receptor, and binds to CD28. Knocking out the genes for such proteins will have multiple consequences—and there may be proteins which redundantly perform some of the same functions, which will compensate for the absence of a knocked-out gene, and make interpretation difficult.
Likewise, actin and microtubules serve structural as well as transport functions: vesicles are transported from the Golgi apparatus to the plasma membrane along microtubules, and actin ruffling and contraction at the synapse periphery may drive pMHC on the APC membrane toward the cSMAC. Disrupting the cytoskeleton will also affect membrane properties (bending and stretching moduli) and thus will have multiple consequences for synapse formation. However, drugs may be introduced in vivo or in vitro, permitting observations in a variety of environments.
Supported membranes have neither the topography nor the composition of the APC membranes with which T cells interact in vivo, and can present only a simplified set of ligands. But a supported lipid membrane containing mobile GPI-linked ligands may provide a good analog to the APC plasma membrane, since the APC cytoskeleton plays a passive role in IS formation.37 In supported membranes, the lipids and GPI-linked proteins exhibit two-dimensional fluidity, which allows interacting T cells to bind and reorganize ligands such as ICAM or pMHC. Moreover, supported membranes may be patterned by a variety of methods on the micron and nanometre scale, which makes them ideal tools for investigation of spatial organization at the immune synapse.
Fixed protein patterns in IS studies
In an exciting recent result, Doh and Irvine found a significant connection between IS geometry and T cell activation in experiments using micropatterned protein surfaces.38 Activation zones of anti-CD3 (which induces T cell clustering) surrounded by adhesion zones of ICAM were photolithographically fabricated to a resolution of 2 µm using biotin–streptavidin affinity to anchor proteins to a photoresist layer. Because photolithography enabled them to pattern an entire face of a flow cell, Doh and Irvine were able to observe proliferation and IL-2 and IFN-γ secretion of T cells interacting with the patterned surface. Although the proteins were immobile and therefore the T cells were not able to reorganize ligands at any length scale, as would be possible with a cell membrane or SLB, the researchers took advantage of this immobility to constrain the geometry encountered by T cells at activation sites. T cells forming synapses with 6 µm diameter anti-CD3 foci stabilized cellular morphology, and proliferated and secreted chemokines in a manner indicating full activation. On annular patterns of anti-CD3, however, T cell morphology remained dynamic or unstable, and the profile of activation as measured by phosphotyrosine and PKC-θ staining and IFN-γ secretion was greatly reduced. Doh and Irvine hypothesize that, for full activation, T cells require a well-defined central TCR cluster corresponding to the cSMAC of the IS.Mobility patterning in IS studies
Some early work with SLB arrays of domains on a relatively large length scale introduced live cells to protein- and photoresist-partitioned bilayers, demonstrating the possibility of spatial control of SLB composition and fluidity as an experimental tool. However, the feature sizes of such patterns were too large for investigation of subcellular phenomena. The first application of micropatterned SLBs to the study of signaling in immune cells introduced rat basophilic leukemia (RBL) mast cells to haptenated SLBs which had been patterned with feature sizes down to 1.5 µm34 by a parylene liftoff process. Fluorescently labeled IgE clearly colocalized with SLB features, and interacting RBL cells showed system-level symptoms of activation: characteristic membrane spreading and ruffling. The potential of this method was more fully realized in a subsequent study39 which used the same cell system and micropatterning technique to investigate three interlinked phenomena: phosphotyrosine signaling, lipid dynamics, and cytoskeletal transport. Cells were transfected with GFP-fused Lyn kinase, GFP-fused lipid raft markers, or GFP-actin, and the spatiotemporal organization was observed over a period of 50 minutes: IgE accumulation on the haptenated features was quickly followed by phosphotyrosine, then Lyn (at 15 minutes), then actin (∼30 minutes), then inner-leaflet raft markers (∼40 minutes). Particularly interesting were the observations that Lyn co-localization with SLB features was not reduced by reduction of phosphotyrosine (which suggests a raft- or cytoskeletal-based mechanism of Lyn transport, and not just binding to phosphotyrosine); and that plasma membrane inner leaflet components redistributed independently of outer leaflet components. These results could not have been obtained without the micropatterned SLBs, because in the absence of subcellular SLB features, no spatial organization of receptors or lipid components would be observable.The advantages of parylene liftoff for SLB patterning include simplicity and a great capacity for production of multiple substrates of large patterned area. Two drawbacks, in the application to the study of subcellular phenomena, are that the feature size is limited to ∼1 µm, and that there are inevitably >2 µm lipid-free gaps between SLB islands. Consequently, cells interacting with the patterned membrane do not encounter a continuous surface—the area covered by SLB islands is significantly smaller than the area of bare substrate. This is not an important issue in the study of FcεRI signaling, because there is no large-scale spatial reorganization in the wild-type system. In the T cell immunological synapse, however, molecular components reorganize on the scale of the entire cell (∼10 µm diameter), and certain features of the IS, such as TCR microclusters and, often, the cSMAC itself, are of the order of 1 µm diameter.
For studying the IS, it is advantageous to have a method of patterning barriers much smaller than the observable features of the system, to minimize perturbation from the physical presence of the barriers themselves and to present a near-continuous membrane to the T cell. To this end, in a recent paper, researchers used electron-beam lithography to define ∼100 nm linewidth designs of chromium on glass substrates, which acted as diffusion barriers to constrain the freedom of motion of lipids and GPI-linked pMHC and ICAM in a supported membrane40(see Fig. 4). T cells interacting with this membrane initiated synapse formation, but barrier constraints on membrane ligands were transferred to the receptors on the T cell membrane, and the mature distribution of ligands and receptors was dictated by substrate geometry (see Fig. 5). ICAM and TCR/pMHC distributions were mutually exclusive, as in the wild-type synapse.
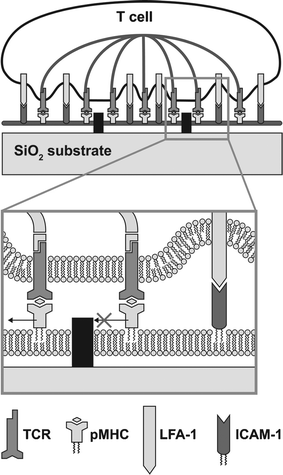 | ||
| Fig. 4 Diagram of a hybrid live T cell-supported membrane junction. Receptors on the cell surface engage cognate ligands in the supported membrane and become subject to constraints on mobility imposed by physical barriers. The cytoskeleton is represented schematically to reflect the active source of central organization observed in our experiments. | ||
 | ||
| Fig. 5 Synapse formation is altered by geometrical constraints of the substrate. T cells were incubated with fluorescently labeled anti-TCR H57 Fab (green) before being introduced to supported bilayers containing GPI-linked pMHC (unlabeled) and ICAM-1 (red). Chromium lines are visible in brightfield, although they are only 100 nm across, verified by electron microscopy. Images are at 10 min after cells were introduced. IS on (A) unpatterned substrate, (B) 2 µm parallel lines, (C) 5 µm square grid, and (D) concentric hexagonal barriers (spacing 1 µm). (E) TCR distribution (grayscale) on 1 µm square grid. (F) Transport map of (E) formed by drawing arrows toward the TCR cluster within the enhanced grid. | ||
The researchers were able to extract information about both the physical mechanism of IS formation and the relationship between spatial organization and the signaling which leads to T cell activation. T cells interacting with a membrane partitioned by a 1 µm grid of chromium lines formed TCR clusters of uniform size, one per grid square, indicating that TCR was not preclustered before T cell contact with the membrane. Each of these TCR clusters was oriented in the corner of its grid square nearest the geometrical center of the synapse, which strongly suggests a centralized, cytoskeletal mechanism of TCR organization, as opposed to a passive process driven by binding, bending, and stretching energies, which Qi et al. put forward as a possibility.30
Interestingly, the partitioned membranes also revealed a mechanism by which T cells use spatial organization at the membrane to regulate internal signaling (see Fig. 6). That this might be the case had been suggested by Lee et al.,32 but in their model the size of clusters was the most significant factor, rather than their radial location in the synapse, as Mossman et al. found. In wild-type synapses, TCR-specific phosphotyrosine signaling (as measured by TCR/pY colocalization), ubiquitous at early time-points up to 2 minutes, was shown to shut off as TCR assembled in the cSMAC by 5 minutes. In synapses frustrated by chromium grids, TCR-specific signaling was strong throughout the interface at early time-points, but, by 5 minutes, signaling had shut off in the geometric center, while remaining relatively high in those TCR clusters arrested in the periphery. Cells forming frustrated synapses exhibited elevated levels of intracellular Ca2+, likely a consequence of activated receptors in the periphery. These results reveal that radial location is a critical parameter in the IS, and that the enzymatic environments in the center and periphery are significantly different.
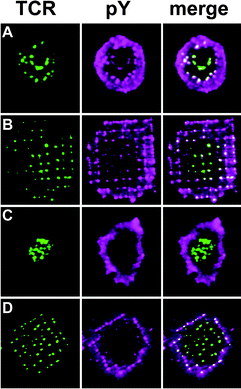 | ||
| Fig. 6 Staining for phosphotyrosine (pY) reveals that TCR-specific signaling is sustained in the periphery of repatterned synapses. T cells, which had been incubated with fluorescently labeled anti-TCR H57 Fab, were allowed to interact with pMHC-ICAM membranes for either 2 or 5 min before being fixed and stained for pY. (A) Synapse on unpatterned membrane at 2 min. TCR clusters are distributed, and relatively enhanced pY staining colocalizes with each cluster. The diffuse ring of pY staining in the periphery is likely associated with cortical actin. (B) Synapse on a 2 µm chromium grid at 2 min. (C) Synapse on an unpatterned membrane at 5 min. (D) Synapse on a 2 µm chromium grid at 5 min. | ||
Future directions
At the single-molecule level, FcεRI and TCR signaling have similarities which could be fruitful to explore. It should be possible to present single or multiple copies of haptens to IgE-loaded mast cells, using micropatterned SLBs—fabricated on the nanometre scale by e-beam lithography—and observe the nucleation of signaling and cytoskeletal networks. Using diffusion barriers, it should also be possible to present T cells with grid squares containing 0 or 1 agonist pMHC (against a background of endogenous pMHC) to observe kinase cascades and cytoskeletal scaffolding in a similar manner. The exact mechanism of TCR transport remains to be determined, and single-molecule observations of TCR trajectories altered by chromium barriers, obtained by TIRF microscopy, will help solve this problem.Also, the recent experiments which revealed spatial regulation of signaling in the IS have left unidentified the kinases and phosphatases responsible. Further immunofluorescent labeling studies are necessary to fill out the picture. On the system level, the importance of the IS in cell development is not well understood, although it is has been found to direct the choice between Th1 and Th2 lineages.41 The hybrid cell/SLB synapse is ideal for representation by computational modeling, especially when modified with geometric partitions. Close collaboration between theorists and experimentalists should result in well-designed experiments to validate hypotheses about the quantitative connection between the synaptic pattern and T cell activation. Because the production of e-beam designs is time-consuming and patterns are restricted to a small area of the substrate, it is essential that a method of fabricating nanoscale SLB patterns over a large area (∼cm2, the area of a flow cell) be developed for experiments to observe long-term effects such as proliferation and cytokine production.
Acknowledgements
The authors thank the National Institutes of Health (USA) for support.References
- D. Bray, M. D. Levin and C. J. Morton-Firth, Nature, 1998, 393, 85 CrossRef CAS.
- E. D. Sheets, D. Holowka and B. Baird, Curr. Opin. Chem. Biol., 1999, 3, 95 CrossRef CAS.
- C. R. F. Monks, B. A. Freiberg, H. Kupfer, N. Sciaky and A. Kupfer, Nature, 1998, 395, 82 CrossRef CAS.
- A. Grakoui, S. K. Bromley, C. Sumen, M. M. Davis, A. S. Shaw, P. M. Allen and M. L. Dustin, Science, 1999, 285, 221 CrossRef CAS.
- E. Sackmann and M. Tanaka, Trends Biotechnol., 2000, 18, 58 CrossRef CAS.
- Y. N. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 551 CrossRef CAS.
- C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Science, 1997, 276, 1425 CrossRef CAS.
- N. Q. Balaban, U. S. Schwarz, D. Riveline, P. Goichberg, G. Tzur, I. Sabanay, D. Mahalu, S. Safran, A. Bershadsky, L. Addadi and B. Geiger, Nat. Cell Biol., 2001, 3, 466 CrossRef CAS.
- E. Hallman, W. R. Burack, A. S. Shaw, M. L. Dustin and P. M. Allen, Immunity, 2002, 16, 839 CrossRef.
- J. C. Stinchcombe, G. Bossi, S. Booth and G. M. Griffiths, Immunity, 2001, 15, 751 CrossRef CAS.
- D. M. Davis, I. Chiu, M. Fassett, G. B. Cohen, O. Mandelboim and J. L. Strominger, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 15062 CrossRef CAS.
- F. D. Batista, D. Iber and M. S. Neuberger, Nature, 2001, 411, 489 CrossRef CAS.
- M. F. Krummel, M. D. Sjaastad, C. Wulfing and M. M. Davis, Science, 2000, 289, 1349 CrossRef CAS.
- T. Yokosuka, K. Sakata-Sogawa, W. Kobayashi, M. Hiroshima, A. Hashimoto-Tane, M. Tokunaga, M. L. Dustin and T. Saito, Nat. Immunol., 2005, 6, 1253 CrossRef CAS.
- K. H. Lee, A. D. Holdorf, M. L. Dustin, A. C. Chan, P. M. Allen and A. S. Shaw, Science, 2002, 295, 1539 CrossRef CAS.
- B. A. Freiberg, H. Kupfer, W. Maslanik, J. Delli, J. Kappler, D. M. Zaller and A. Kupfer, Nat. Immunol., 2002, 3, 911 CrossRef CAS.
- M. Tanaka and E. Sackmann, Nature, 2005, 437, 656 CrossRef CAS.
- H. M. McConnell, T. H. Watts, R. M. Weis and A. A. Brian, Biochim. Biophys. Acta, 1986, 864, 95 CAS.
- T. M. Bayerl and M. Bloom, Biophys. J., 1990, 58, 357 Search PubMed.
- J. T. Groves and M. L. Dustin, J. Immunol. Methods, 2003, 278, 19 CrossRef CAS.
- A. A. Brian and H. M. McConnell, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 6159 CAS.
- T. H. Watts, H. E. Gaub and H. M. McConnell, Nature, 1986, 320, 179 CrossRef CAS.
- A. Tozeren, K. L. Sung, L. A. Sung, M. L. Dustin, P. Y. Chan, T. A. Springer and S. Chien, J. Cell Biol., 1992, 116, 997 CrossRef CAS.
- S. Pautot, H. Lee, E. Y. Isacoff and J. T. Groves, Nat. Chem. Biol., 2005, 1, 283 CrossRef CAS.
- D. G. Hafeman, V. V. Tscharner and H. M. McConnell, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 4552 CrossRef CAS.
- D. Holowka and B. Baird, Semin. Immunol., 2001, 13, 99 CrossRef CAS.
- W. C. Moss, D. J. Irvine, M. M. Davis and M. F. Krummel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15024 CrossRef CAS.
- R. E. Dolmetsch, K. L. Xu and R. S. Lewis, Nature, 1998, 392, 933 CrossRef CAS.
- C. Utzny, M. Faroudi and S. Valitutti, Biophys. J., 2005, 88, 1 CAS.
- S. Y. Qi, J. T. Groves and A. K. Chakraborty, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6548 CrossRef CAS.
- S.-J. E. Lee, Y. Hori and A. K. Chakraborty, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4383 CrossRef CAS.
- K. H. Lee, A. R. Dinner, C. Tu, G. Campi, S. Raychaudhuri, R. Varma, T. N. Sims, W. R. Burack, H. Wu, O. Kanagawa, M. Markiewicz, P. M. Allen, M. L. Dustin, A. K. Chakraborty and A. S. Shaw, Science, 2003, 302, 1218 CrossRef CAS.
- E. Sackmann, Science, 1996, 271, 43 CrossRef CAS.
- R. N. Orth, M. Wu, D. A. Holowka, H. G. Craighead and B. A. Baird, Langmuir, 2003, 19, 1599 CrossRef.
- J. T. Groves, N. Ulman and S. G. Boxer, Science, 1997, 275, 651 CrossRef.
- J. T. Groves and S. G. Boxer, Acc. Chem. Res., 2002, 35, 149 CrossRef CAS.
- C. Wulfing and M. M. Davis, Science, 1998, 282, 2266 CrossRef CAS.
- J. Doh and D. J. Irvine, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5700 CrossRef CAS.
- M. Wu, D. Holowka, H. G. Craighead and B. Baird, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13798 CrossRef CAS.
- K. D. Mossman, G. Campi, J. T. Groves and M. L. Dustin, Science, 2005, 310, 1191 CrossRef CAS.
- R. A. Maldonado, D. J. Irvine, R. Schreiber and L. H. Glimcher, Nature, 2004, 431, 527 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |