Ring closing enyne metathesis : A powerful tool for the synthesis of heterocycles
Hélène
Villar
ab,
Marcus
Frings
a and
Carsten
Bolm
*a
aInstitut für Organische Chemie der RWTH Aachen, Landoltweg 1, 52074 Aachen, Germany. E-mail: Carsten.Bolm@oc.rwth-aachen.de
bNew address: Equipe Matériaux Tensioactifs, BP 239, Polymères et Colloïdaux, 54506 Vandœuvre-lès-Nancy, UMR 7565 CNRS-UHP, Université Henri Poincaré Nancy I, France
First published on 4th October 2006
Abstract
The ring closing metathesis (RCM) is a powerful method in organic synthesis for the preparation of cyclic compounds by formation of new carbon–carbon bonds. In the past years a particular subclass of the RCM, the ring closing enyne metathesis (RCEYM), has attracted attention due to its synthetic potential in the generation of ring structures with 1,3-diene moieties, which can subsequently be further functionalised. In this tutorial review mechanistic considerations will be described and the synthetic power of this useful and attractive carbon–carbon bond forming reaction will be illustrated by recent examples of RCEYM applications in the preparation of heterocyclic compounds .
 Hélène Villar | Hélène Villar studied chemistry at the University PARIS XI in Orsay (France). In 2003 she finished her doctoral work with Dr Guibé in Orsay on the mechanistic study of 3-exo-trig radical cyclisation induced by samarium diiodide. She obtained an Alexander von Humboldt postdoctoral fellowship to join the group of Professor Bolm at RWTH Aachen University (Germany), where she worked on synthesis of heterocyclic sulfoximines by ring closing metathesis. Since 2005 she has been a lecturer in Professor Gérardin's group at the Henri Poincaré University in Nancy (France), studying conception, synthesis and properties of polyfunctionalised perfluorinated amphiphilic compounds. |
 Marcus Frings | Marcus Frings was born in Eschweiler (Germany) and studied chemistry at the RWTH Aachen University (Germany). In 2005 he completed his diploma thesis under the supervision of Professor Bolm in which he investigated the application of sulfoximines in ring closing metathesis. Currently, he is a doctoral student in the group of Professor Bolm. His project is focused on methodology development and the synthesis of sulfoximine-containing biologically active compounds. |
 Carsten Bolm | Carsten Bolm studied chemistry at the TU Braunschweig in Germany and at the University of Wisconsin in Madison (USA). In 1987 he finished his doctoral work with Professor Reetz in Marburg (Germany). After postdoctoral studies at MIT, Cambridge (USA), with Professor Sharpless, Carsten Bolm began to work on his habilitation in Basel (Switzerland) in the group of Professor Giese. In 1993 he became Professor of Organic Chemistry at the University of Marburg (Germany), and since 1996 he has been a full professor for Organic Chemistry at the RWTH Aachen University (Germany). He has had visiting professorships at the universities in Madison, Wisconsin (USA), Paris (France), Florence (Italy), Milan (Italy), Namur (Belgium) and Tokyo (Japan). His list of awards includes the Heinz–Maier–Leibnitz prize, the ADUC-Jahrespreis for habilitands, the annual prize for Chemistry of the Akademie der Wissenschaften zu Göttingen, the Otto-Klung prize, the Otto-Bayer award, a fellowship of the Japan Society for the Promotion of Science and the Prix Franco-Allemand by the Société Francaise de Chimie. |
1. Introduction
The Nobel Prize in Chemistry 2005 was equally shared by Yves Chauvin (Institut Français du Pétrole, Rueil-Malmaison, France), Robert H. Grubbs (California Institute of Technology, Pasadena, USA) and Richard R. Schrock (Massachusetts Institute of Technology, Cambridge, USA) for their outstanding contributions in the area of olefin metathesis reactions.1 Chauvin had investigated the mechanism of this unusual carbon–carbon-bond forming reaction, and studies by Grubbs and Schrock have led to synthetically highly efficient ruthenium, molybdenum, and tungsten metathesis catalysts. As apparent from today's textbooks, their discoveries had a tremendous impact on modern organic chemistry and both academia and industry largely benefited from their findings.Metathesis , with its multiple facets, has become one of the most important chemical transformations.2–7 Depending on the type of unsaturated bond involved in the metathesis process, three major categories of olefin metathesis can be distinguished: diene, enyne and diyne metathesis. The structural change, which occurs during the process, can be further subdivided into ring closing, ring opening, and cross metathesis. Here, we will focus on one of these classes, which has recently been shown to be a highly powerful method for the generation of ring structures from functionalised molecules with tethered alkenes and alkynes: the ring closing enyne metathesis .
The first reaction of this type was reported by Katz and Sivavec in 1985, who used a Fischer tungsten carbene complex in an intramolecular enyne metathesis giving an all-carbon product in moderate (31%) yield.8 Later, analogous transformations have been performed with molybdenum and chromium carbene complexes. Subsequently, the development of well-defined ruthenium carbene catalysts with a wide functional group tolerance has considerably improved the selectivity and scope of RCEYM reactions. The most common catalysts are presented in Fig. 1.
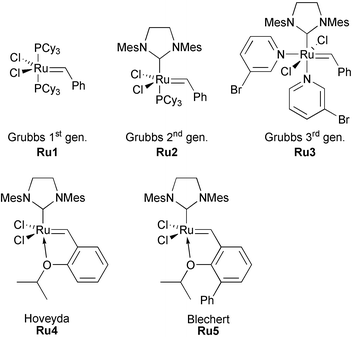 | ||
| Fig. 1 Common ruthenium carbene complexes used in metathesis reactions. | ||
Grubbs’ tremendous accomplishments resulted in three different generations of ruthenium metathesis complexes, Ru1, Ru2 and Ru3.9 Hoveyda's and Blechert's ruthenium complexes Ru410 and Ru511 can be considered as derivatives of the Grubbs catalyst family bearing loosely chelating groups.12
2 Mechanism outlines
Despite the fact that enyne metathesis is closely related to alkene metathesis, the mechanism of the former is by far less understood than the latter metathesis reaction. Formally, in a RCEYM reaction a carbon–carbon bond formation between an olefinic and an alkynylic carbon occurs affording a cyclic 1,3-diene. The process is complex and involves a number of steps.Two mechanisms for enyne metathesis reactions have been discussed in the literature: first, a metal salt-catalysed enyne bond reorganization, and, second, a metal carbene-mediated enyne metathesis reaction. After a short presentation of both mechanisms, we will focus on transformations following the second mechanistic pathway.
2.1 Metal salt-catalyzed enyne bond reorganisation
Trost pioneered the employment of late transition metal salts such as Pd(II) for triggering enyne bond reorganisation.13,14 The reaction starts by a bidentate coordination of enyne 1 to the metal salt generating 2, followed by oxidative cyclisation to give metalacyclopentene 3. Next, reductive elimination occurs to produce cyclobutene4. The process is completed by bond isomerisation and ring opening of 4 to afford diene 5. Scheme 1 illustrates a generic pathway of such ‘metal-templated enyne bond reorganisation’.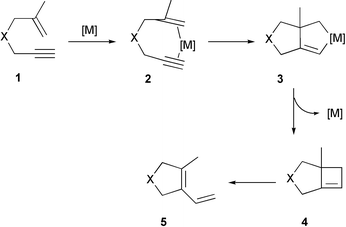 | ||
| Scheme 1 Metal-templated enyne bond reorganisation. | ||
Depending on the nature of catalyst, a second pathway can be considered.15 For Pt(II) Fürstner proposed a mechanism involving a π-alkyne activation by the metal (Scheme 2).16
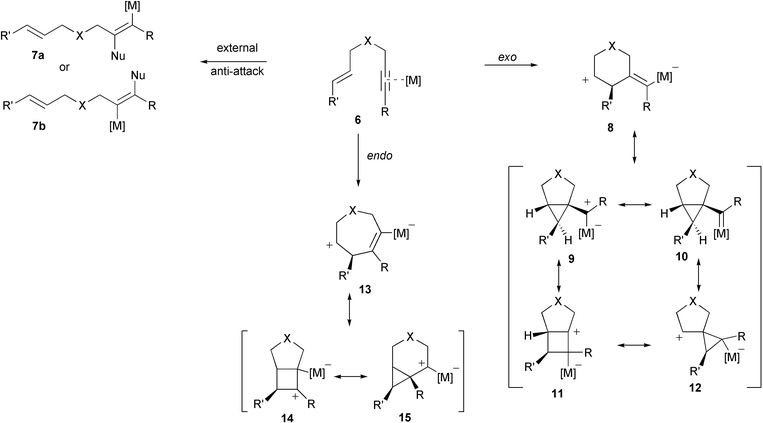 | ||
| Scheme 2 General representation of metal-assisted alkyne activation. | ||
On one hand, an intermolecular attack of the metal-coordinated alkyne 6 by an external nucleophile in an anti-manner can lead to open-chain products 7a and 7b. If this attack occurs in an intramolecular (exo- or endo-) fashion, metallated intermediates 8–12 and 13–15 result, which represent canonical forms of a non-classical homoallyl-cyclopropylmethyl-cyclobutyl cation.
This concept was further elaborated by Echavarren, who concluded on basis of DFT calculation results that ‘the similarities between the intramolecular reactions of alkynes with alkene catalyzed by late transition metal complexes and the carbocationic rearrangements of the cyclopropyl-methyl-cyclobutyl manifold, pointed out by Fürstner, is remarkable. However, differences undoubtedly exist due to the metal stabilization of reactive species.17
2.2 Metal carbene-mediated enyne metathesis
The ruthenium carbenes developed by Grubbs reveal an excellent catalytic efficiency in enyne metathesis reactions, and their application in synthesis is attractive because they possess a remarkable functional group tolerance. Several aspects of the RCEYM mechanisms have been questioned, and either they lack or have incomplete answers.18,19 For example: (1) Which of the multiple bonds, the double or triple bond, reacts first with a ruthenium carbene complex? (2) What is the effect of the ring size on the regioselectivity? (3) What is the role of ethylene if used as reaction atmosphere?Possible reaction courses are shown in Schemes 3 and 4. If the RCEYM proceeds by initial reaction of the alkynylic part of enyne 1 with the ruthenium carbene complex (Ru), the sequence of events is called ‘yne-then-ene pathway’ (Scheme 3). Two alternative reaction pathways (entitled a-exo and a-endo) can then be envisaged. On one hand, the metal side of the ruthenium carbene complex (Ru) can combine with the internal carbon of the alkyne of 1 forming ruthenacyclobutene 16 (Scheme 3, left; a-exo pathway). Ring-opening of 16 leads to vinylic ruthenium carbene complex 17, and subsequent intramolecular [2 + 2] cycloaddition affords ruthenacyclobutane 18. Consequently, upon ring-opening of 18exo product 5 is formed. On the other hand, when the metal center of the ruthenium carbene complex (Ru) combines with the terminal carbon of the alkyne part of 1, ruthenacyclobutene 19 results, which upon ring opening is converted into ruthenium carbene complex 20. When 20 then reacts intramolecularly with its terminal olefinic part by [2 + 2] cycloaddition , formation of ruthenacyclobutane 21 results, which finally leads to the formation of endo product 22. On both routes the metal carbene catalyst Ru is regenerated in the last step.
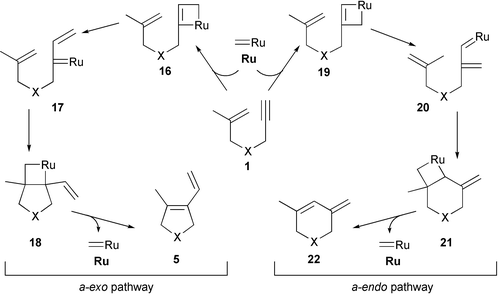 | ||
| Scheme 3 ‘Yne-then-ene pathway’ of RCEYM reactions. | ||
 | ||
| Scheme 4 ‘Ene-then-yne pathway’ of RCEYM reactions. | ||
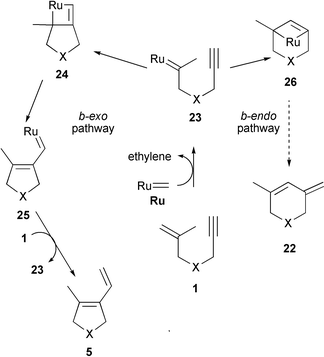
On a so-called ‘ene-then-yne pathway’ (Scheme 4) the ruthenium carbene catalyst (Ru) first reacts with the olefinic moiety of 1 to produce alkylidene 23. Then again, two possible event sequences can be distinguished. The b-exo pathway (Scheme 4, left) involves the ring closure of 23 to give ruthenacyclobutene 24 followed by fragmentation of the 4-membered ring to afford vinyl carbene25. Subsequent reaction with a second equivalent of enyne 1 leads to (exo product) 5 and regenerates alkylidene 23 for the next catalytic cycle. Alternatively, the b-endo pathway involves the formation of bicyclic ruthenacyclobutene 26 resulting from the combination of the ruthenium side of 23 with the internal carbon of the triple bond. Ring-opening then leads to (endo product) 22. Assuming that the intermediacy of the highly strained ruthenacyclobutene 26 is unlikely, one would expect the ‘ene-then-yne process’ to result in the formation of exo product 5 with high preference.
With respect to the competition between the ‘yne-then-ene’ and ‘ene-then-yne’ pathways, Mori noted:20 ‘We have already reported that in the enyne metathesis, ruthenium carbene complex [Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ] should react at first with the alkyne part’ (faster reaction rate of RuCH2 with the alkyne moiety compared to transalkylidenation with the alkene moiety). ‘However, the reaction rates of diene metathesis and enyne metathesis were almost the same.’
] should react at first with the alkyne part’ (faster reaction rate of RuCH2 with the alkyne moiety compared to transalkylidenation with the alkene moiety). ‘However, the reaction rates of diene metathesis and enyne metathesis were almost the same.’
On the basis of NMR experiments and substrate/catalyst concentration studies in the synthesis of the vinylic lactone differolide (31), Hoye favored the ‘ene-then-yne’ mechanism (Scheme 5, right).21 Reactions monitored by NMR showed the rapid appearance of alkylidene 32, which suggested that the initiation by transalkylidenation was fast relative to the carbene enyne cycloaddition . Despite this indicative evidence for the ‘ene-then-yne’ mechanism, the ‘yne-then-ene pathway’ could not unequivocally be ruled out finally.
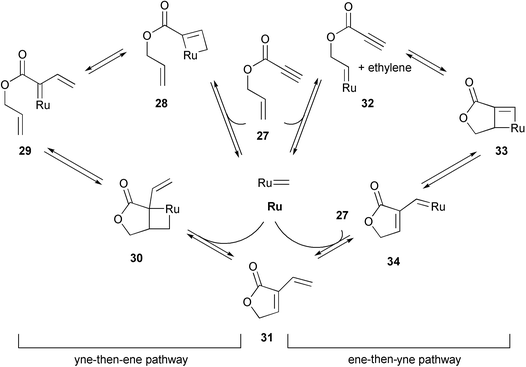 | ||
| Scheme 5 Mechanistic pathways towards vinylic lactone 31 investigated by NMR spectroscopy. | ||
Enyne metathesis reactions with substrates having sterically demanding alkyne portions indicated a preference for the ‘yne-then-ene pathway’.22a Contrary results, however, could be deduced from other studies.22b
Another important issue in the enyne metathesis mechanism is the exo/endo selectivity, since it dictates the ring size of the resulting product. As already shown in Schemes 3 and 4 the endo-mode ring closure products have one carbon more in the ring than those derived from the exo-mode closure. Interestingly, in formations of small- to medium-sized rings the RCEYM reaction generally follows the exo-mode pathway, whereas macrocycles are commonly obtained by the endo-mode. Hansen and Lee noted in this context that the ‘ene-then-yne’ route explains the observed change in exo/endo selectivity in a more reasonable and straightforward way than the ‘yne-then-ene’ route.23 In the ‘ene-then-yne pathways’ (Scheme 4) the exo/endo mode selectivity appears to be a direct consequence of the ring strain associated with the respective ruthenacyclobutene intermediates, and the tether length dictates the reaction course. Following the ‘yne-then-ene’ pathways (Scheme 3), the switch from the exo to the endo selectivity (when forming small or large rings, respectively) would be more difficult to explain.
In 1998, Mori investigated the effect of ethylene gas on RCEYM reactions.18,20 It was found that in conversions of substrates with terminal alkynyl groups the yields could significantly be increased, when the reactions were carried out under an atmosphere of ethylene. In contrast, reactions of substrates with non-terminal alkynyl groups were not affected. Mechanistically the activity increase in the presence of ethylene was explained by a constant ‘reactivation’ of the ruthenium catalyst keeping it in an active state through formation of ruthenacyclobutane 38 (Scheme 6). Under standard reaction conditions in the absence of ethylene, the ruthenium catalyst is trapped in less active species 39 and 40 (derived from the RCEYM product and ruthenium alkylidenes). Thus by shifting the equilibrium towards intermediate 38 the active catalyst can readily react with further starting material 35.
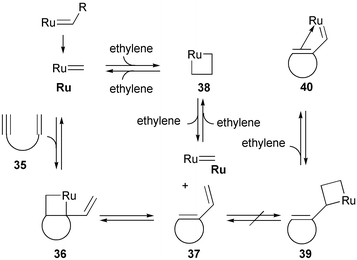 | ||
| Scheme 6 RCEYM reactions in the presence of ethylene. | ||
Recently, the observed rate enhancement by ethylene has been investigated in more detail and from isotopic labelling studies Lloyd-Jones deduced evidence of an ‘ene-then-yne’ pathway with the involvement of a second catalytic cycle.24
The beneficial effect of ethylene in RCEYM reactions giving small- to medium-sized rings and nitrogen or oxygen heterocycles appears to be general. In the formation of large-membered rings Hansen and Lee recently showed that the use of an ethylene atmosphere leads to a competitive cross metathesis (CM) of the alkyne moiety with ethylene, which is presumably due to the relative slow rate of macrocyclization via enyne metathesis .23 As a result, triene 42 is formed (Scheme 7), which can serve as new substrate for a subsequent diene RCM. In this later process, the catalyst reacts first with the isolated double bond to form ruthenium alkylidene 43, which then undergoes ring closure with the distal monosubstituted double bond of the 1,3-diene moiety affording selectively endo-product 44.
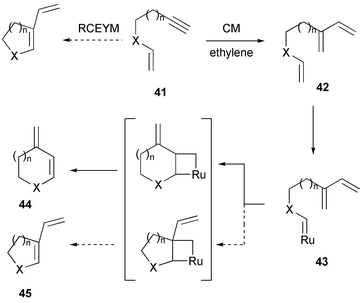 | ||
| Scheme 7 Cross metathesis reaction followed by ring closure in the presence of ethylene. | ||
A positive effect of an ethylene atmosphere on the yield of enyne metathesis products has also been observed in cyclisations of carbohydrate-derived enynes giving polyhydroxylated 1-vinylcycloalkenes.25
Mori demonstrated the importance of substituents on the olefinic part of the enyne on the RCEYM reaction (Scheme 8).26 The first example involves 1,1-disubstituted olefins 46a–c having a terminal triple bond. In reactions under ethylene atmosphere exclusively exo compounds 47a–c were formed. The relatively low yield of 47a was proposed to be a consequence of the high reactivity of the dienyl moiety. In products 47b and 47c this part is more shielded by bulky substituents, and those compounds are therefore less prone to undergo subsequent reactions involving the dienyl fragments.
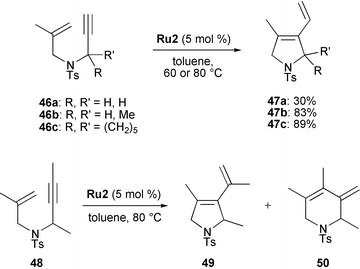 | ||
| Scheme 8 RCEYM starting from substituted enynes. | ||
RCEYM reactions of enyne 48 bearing a 1,1-disubstituted olefinic and an internal alkynylic group have also been studied. In this case, products 49 and 50 stemming from the exo- and endo-pathways, respectively, were obtained as an inseparable mixture. The authors write26b ‘The reason why RCM of enyne having a monosubstituted alkene or a terminal alkyne gave only the exo compound, while that of enyne having a disubstituted alkene and an internal alkyne gave a mixture of both exo and endo products is not clear. Presumably, the steric effect on the multiple bond affected the ring size of the product and their ratio.’
3. Examples of heterocycle syntheses by RCEYM
Enyne metathesis is a powerful carbon–carbon bond-forming process, which leads to 1,3-dienes from alkenes and alkynes. Despite their obvious synthetic value, RCEYM reactions have by far less been used in targeted synthesis than their well-established diene metathesis counterpart. Since many synthetic applications of RCEYM reactions have already been covered in the excellent review by Diver and Giessert,2a the overview here will only detail examples relating to the syntheses of heterocycles with one or more heteroatoms taken from reports published after 2004.An efficient route to 4/x/6 (with x = 5–7) polycyclic β-lactams was described by Genêt in 2004.27 The 4/x fused-bicyclic frameworks were obtained by RCEYM, and subsequent Diels–Alder reactions afforded the 4/x/6 annulated β-lactam systems (Scheme 9). These so-called ‘tricyclic carbapenems’ are of great synthetic interest due to their enhanced stability and activity against resistant bacteria. Also noteworthy is that the 4/5 annulated skeleton—albeit generally difficult to prepare—is a highly attractive target, since it is part of the framework of the biologically active Sanfetrinem.
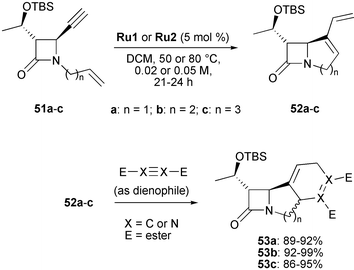 | ||
| Scheme 9 RCEYM followed by Diels–Alder reaction to afford 4/x/6 annulated β-lactams. | ||
Application of Grubbs' first-generation catalyst Ru1 in dichloromethane (DCM) at 50–80 °C in sealed tubes gave bicyclic lactams 52b and 52c (4/6 and 4/7 ring systems) in very good yields (87% and 75%, respectively) within 21–24 h (Table 1, entries 3 and 5). In contrast, the 4/5 annulated product 52a was obtained in only 29% yield and starting material could be recovered (entry 1). Using Grubbs' second-generation catalyst Ru2 gave consistantly high yields for all products. The authors explained the reactivity difference between 52b and 52c on one hand and 52a on the other by the formation of highly strained intermediates leading to a thermodynamically unfavourable partial loss of resonance in the lactam function. In reactions with 52a this aspect is particularly important as indicated by 13C-NMR spectroscopy and molecular modelling. Compared to enyne 51a and products 52b and 52c, the carbonyl group of the 4/5 bicyclic lactam 52a shows a much higher chemical shift value (δ = 180.1 ppm). Furthermore, molecular modelling revealed that in compound 52a the deformation angle between the C–N bond of the newly formed 5-membered ring and the C–N bond of the 4-membered lactam ring was 54°, whereas lower values were found for the respective angles of enyne 51a (29°) and bicycles 52b and 52c (both 42°). The resulting strong decrease of resonance in 52a is thermodynamically unfavourable explaining why the more active second-generation catalyst was needed to form the 4/5 system of 52a in good yield. Finally, treatment of dienes 52a–c with dienophiles such as maleimide and dimethylacetylenedicarboxylate (DMAD) gave tricyclic carbapenems 53a–c in excellent yields. The diastereoselectivities in these cycloadditions were moderate, and only when 4-phenyl-1,2,4-triazoline-3,5-dione (PTAD) was used as dienophile a single diastereomer was obtained.
The synthesis of cyclic 1,2-diaza cycloalkenes by RCEYM was recently described by Tae and Hahn (Scheme 10).28 It was the first time that enynes, which were tethered by an N–N bond, were employed in RCEYM reactions to form 6-, 7-, and 8-membered cyclic hydrazines. These molecules are of great synthetic value, since several biologically active compounds contain cyclic skeletons with N–N bonds, and a general and versatile approach towards such compounds was still lacking.
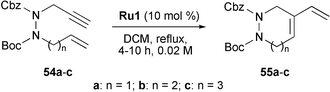 | ||
| Scheme 10 RCEYM in the synthesis of cyclic hydrazines. | ||
The starting materials were enynes 54a–c (n = 1–3), which could be obtained under standard conditions from 1-tert-butoxycarbonyl-2-carbobenzyloxyhydrazine and the appropriate bromoalkenes and propargyl bromide. They were then cyclised by RCEYM using 10 mol% of Grubbs' first-generation catalyst Ru1 in refluxing DCM. With a substrate concentration of 0.02 M, the 6-membered hydrazine-based heterocycle 55a(n = 1) was obtained in excellent yield (99%) after 4 hours. The syntheses of 7- and 8-membered cyclic hydrazines55b (n = 2) and 55c (n = 3) required longer reaction times and those products were isolated in 70% yield each after 8 and 10 h, respectively.
The reactivity of 1,2-diaza compounds 55a–c was then investigated in Diels–Alder cycloadditions with DMAD as dienophile. Subsequent DDQ-oxidation of the products afforded bicyclic aromatic heterocycles in very good yields.
Kim and Lee also synthesised cyclic 1,2-diaza cycloalkenes by RCEYM and demonstrated the value of the process for the synthesis of macrocyclic amides (Scheme 11).29 In their case, functionalised hydrazine derivatives such as 56 and 59 containing keto or ester groups in the alkenyl or alkynyl chain were used. Treatment of these substrates with 5 mol% of Grubbs' second-generation catalyst Ru2 in a 0.002 M solution of refluxing DCM under a continuous flow of ethylene yielded 8- and 13-membered cyclic hydrazines57 and 60. Noteworthy is the fact that the cyclisation of enyne 56 led to a 1:1 mixture of RCM product 57 and the corresponding ethylene CM product 58. The reaction of enyne 59 afforded a 2 : 1 : 1 mixture of 1,3-diene 60 and two ethylene CM products 61a and 61b. As discussed earlier in the mechanistic part of this review, formation of macrocycles favours the endo-pathway. Thus it is not surprising that in the RCEYM reaction of 59 only the endo-cyclic 13-membered product 60 was obtained. In contrast, cyclisation of 56 afforded exo-cyclic 8-membered product 57.
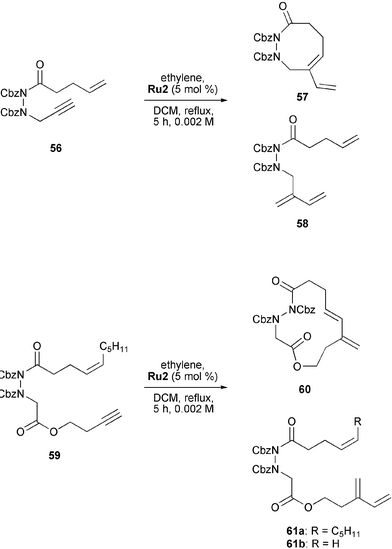 | ||
| Scheme 11 RCEYM reactions of functionalised enynes leading to cyclic hydrazine derivatives. | ||
Snapper synthesised alkenyl cyclopropanes through a tandem RCEYM–cyclopropanation sequence (Scheme 12).30 Treatment of the enynes 62 (n = 1–3) with 10 mol% of Grubbs' first-generation catalyst Ru1 under an atmosphere of ethylene produced the 5-, 6- and 7-membered ring closure products 63 which were directly subjected to cyclopropanation by adding various diazo compounds (R1 = H or CO2Me, R2 = CO2Me, CO2Et, CO2t-Bu or TMS) to give vinyl cyclopropanes 64 in good yields (34–71%). In this reaction alkylidene Ru1 serves as catalyst for both RCEYM and cyclopropanation. However, this tandem sequence seems to be specific to Grubbs' first-generation catalyst Ru1. With the second-generation catalyst Ru2 only dimer 65 was formed and no cyclopropanation product could be observed. Furthermore, Ru1 led to a high regioselectivity in the cyclopropanation step with cyclopropanation occurring almost exclusively on the less hindered olefin. In contrast, Dixneuf discovered that utilising Cp*RuCl(cod) as catalyst compound 66 (R = Me) with opposite regioselectivity was formed.31
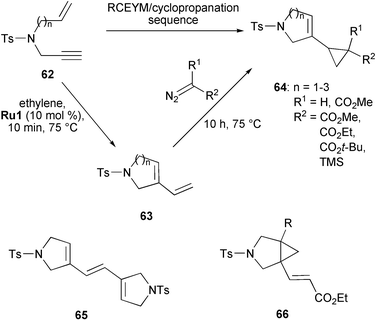 | ||
| Scheme 12 Synthesis of alkenyl cyclopropanes by tandem RCEYM–cyclopropanation with high regioselectivity. | ||
Gracias reported on the synthesis of 5/6-, 5/7- and 5/8-fused imidazo azepine derivatives by RCEYM reactions (Scheme 13).32 The starting imidazoles, such as compound 67, were prepared from tosylmethyl isocyanides, primary amines and aldehydes or from secondary amino aldehydes and amino esters by the van Leusen reaction. Noteworthy is that for the enyne ring closure it was necessary to pretreat the imidazoles with an equivalent of p-TsOH to form the corresponding imidazolium ions. This treatment prevented the lone pair of the imidazole nitrogen from inactivating the metathesis catalyst Ru2. Without p-TsOH no reaction was observed. The RCEYM reactions were then carried out in refluxing DCM with 10 mol% of Grubbs' second-generation catalyst Ru2 to give the fused bicyclic enyne products in good yields (54–82%). The best result (82% yield) was obtained in formation of the 5/7-fused heterocycle 68. With terminal alkynes only yields of less than 5% were observed for the RCEYM products.
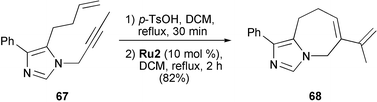 | ||
| Scheme 13 Synthesis of fused bicyclic imidazole derivatives by RCEYM. | ||
Brown synthesised 7-membered cyclic sulfamides 70a–c by RCEYM starting from sulfamide-linked enynes 69a–c (Scheme 14).33 Whereas the reaction was sluggishly at room temperature, it proceeded well and rapidly when carried out under microwave irradiation (MW) at 100 °C in a sealed system. Thus, treatment of enynes 69a–c with 3 mol% of Grubbs' second-generation catalyst Ru2 gave heterocycles 70a–c in good yields (68–81%).
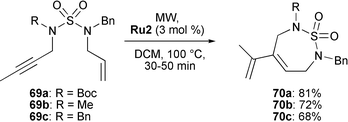 | ||
| Scheme 14 Synthesis of cyclic sulfamides by RCYEM. | ||
Interestingly, RCEYM reactions of the corresponding terminal alkynes 71 proceeded differently and gave three major products. Their ratio depended on the reaction conditions (Table 2). Use of low catalyst loadings of Ru2 in the reactions of enynes 71b and 71c afforded the expected RCEYM products 72b and 72c, respectively (Table 2, entries 2, 3, 5 and 6). In contrast, N-Boc-protected substrate 71a favoured the formation of RCEYM–homo-CM product 73a (Table 2, entry 1). If the amount of catalyst Ru2 was increased to 20 mol%, also enynes 71b and 71c formed the corresponding RCEYM–homo-CM products 73b and 73c in strong preference over the regular RCEYM heterocycles (Table 2, entries 4 and 7). Furthermore, by-products 74a–c were obtained in all reactions resulting from CM of 72 with the benzylidene group of the catalyst.
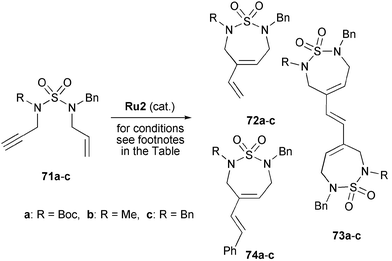
Inspired by the formation of CM products 74, a one-pot RCEYM–CM sequence was developed. Thus, when the reactions of terminal alkynes 71a–c were carried out in the presence of styrene or methyl acrylate, the desired RCEYM–CM products (analogous to 74; not shown) were formed in good yields with predominant E-selectivity.33
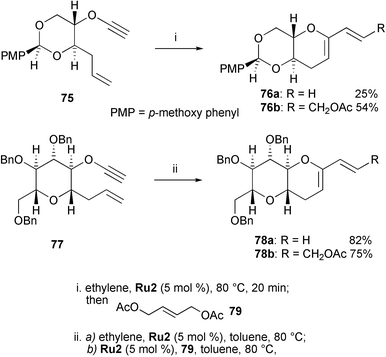
Building blocks for polyethers are interesting targets for RCEYM reactions since they can often be found in marine natural products such as gambierol or hemibrevetoxin B. In this context, Clark reported the synthesis of 6-membered cyclic ethers, which can either be obtained by a one-pot RCEYM–CM reaction or, alternatively, by a ring construction followed by a side chain introduction (Scheme 15).34 For example, treatment of enyne 75 with Grubbs' second-generation catalyst Ru2 at 80 °C in toluene under an atmosphere of ethylene gave cyclic ether 76a (R = H) to which (E)-2-butene-1,4-diol diacetate (79) was added whilst purging with argon. After 16 h at 70 °C CM product 76b (R = CH2OAc) was obtained in 54% yield, along with 25% yield of the regular RCEYM 1,3-diene 76a. For this one-pot procedure a careful adjustment of the enyne concentration was important. Thus, formation of 1,3-diene 76a and almost no CM product was observed when the concentration of 75 was higher than 1 M or lower than 0.01 M. When enyne 77 was subjected to the RCEYM reaction, bicyclic heterocycle 78a (R = H) was isolated in 82% yield. Again, this product was further functionalised by CM to give disubstituted olefin 78b (R = CH2OAc) in 75% yield. An attractive feature of this RCEYM–CM sequence is that complex side chain functionalities can easily be introduced.
 | ||
| Scheme 15 Synthesis of cyclic ethers by RCEYM followed by CM. | ||
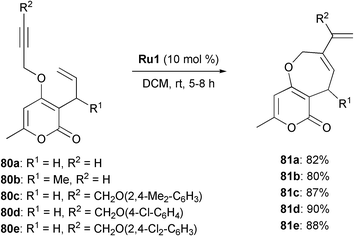
Fused pyrone derivatives are widely present in physiologically active substances but only very little is known about medium ring oxacycle fused pyrones. This is due to a lack of general syntheses of these ring systems. Recently, Majumdar described an approach towards the building of oxepin-annulated pyrones by RCEYM (Scheme 16).35 The bicyclic compounds 81a–e were prepared with very good yields (80–90%) from enynes 80a–e in DCM at room temperature in the presence of 10 mol% Grubbs' first-generation catalyst Ru1. Again, utilising terminal alkynes like 80a or 80b as substrates resulted in slightly lower yields (82% and 80%, respectively) compared to the substituted propargyl aryl ethers 80c–e which gave the metathesis products in better yields (87–90%). The obtained heterocycles 81c–e were then subjected to Diels–Alder cycloadditions and tricyclic pyranoxepin derivatives were formed as single diastereomers in excellent yields (95% and 96%, respectively).
 | ||
| Scheme 16 Synthesis of oxepin-annulated pyrone derivatives by RCEYM. | ||
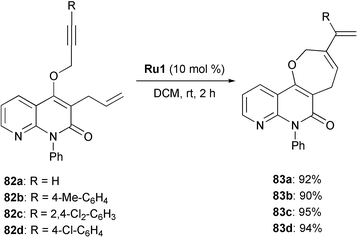
In the same context Majumdar reported the synthesis of oxacycle-annulated 1,8-naphthyridinones by RCEYM which are of particular interest due to the broad spectrum of biological activities of substituted naphthyridine derivatives (Scheme 17).36 Enynes 82a–d were treated with 10 mol% of metathesis catalyst Ru1 in DCM at room temperature and ring closure proceeded smoothly to afford the oxepine derivatives 83a–d in very high yields (90–95%).
 | ||
| Scheme 17 Synthesis of tricyclic 1,8-naphthyridinones derivatives by RCEYM. | ||
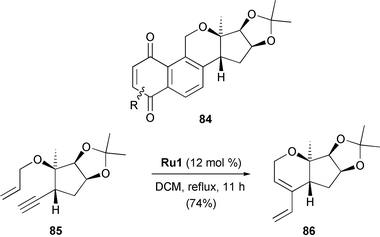
Kaliappan synthesised sugar–oxasteroid–quinone hybrid structures 84, featuring RCEYM reactions as key steps (Scheme 18).37 Using sugar-derived enyne 85 and Grubbs' first-generation catalyst Ru1 in refluxing DCM led to 1,3-diene 86 in 74% yield. By treatment with dienophiles, 86 was then converted into the corresponding Diels–Alder cycloadducts which underwent self-aromatization/oxidation on silica gel to give the target products 84. Thus, RCEYM as key reaction offers a versatile and general route to interesting hybrid molecules with three different structural motifs, which may exhibit significant biological activity.
 | ||
| Scheme 18 Synthesis of sugar–oxasteroid–quinone hybrid structures by RCEYM. | ||
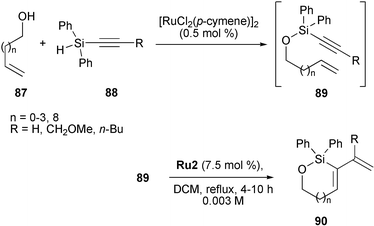
Although RCEYM reactions are known to tolerate heteroatoms such as B, N, O, P and S in the alkynylic part of the enyne, Si-substituted heterocycles derived from alkynyl silyloxy-tethered enynes were not reported until 2004, when Lee described the first siloxacycles 90 prepared by RCEYM (Scheme 19).38 Enynes 89 were easily prepared by ruthenium catalysis starting from alkenylic alcohols87 and alkynylsilanes 88. Filtration through a pad of silica gel removed the first ruthenium catalyst {[RuCl2(p-cymene)]2} and subsequent treatment of the resulting silyl ethers 89 with Grubbs' second-generation catalyst Ru2 in refluxing DCM gave siloxacycles 90 in yields ranging from 30–85%. In this manner small- and medium-sized (5–9-membered) heterocycles as well as 13-membered macrocycles containing silicon as heteroelement were accessible. Interestingly, when the first ruthenium catalyst was not removed and the resulting siloxy-tethered enyne directly subjected to the RCEYM, the yield of 1,3-dienylvinylsilane 90 was significantly lower. Furthermore the alkynylsilyloxy tether appeared to activate the substrates for RCEYM, as indicated by the fact that the corresponding all carbon-tethered enynes (not shown here) did not undergo ring closure under identical conditions.
 | ||
| Scheme 19 RCEYM in the synthesis of cyclic 1,3-dienylvinylsilanes. | ||
All heterocycles 90 were exo products and interestingly also silyl ether 89 with n = 8 favoured the formation of an exo 13-membered heterocycle 90 despite the fact that this tether length usually preferred endo ring closures. As explanation the authors suggested that the metathesis process was initiated on the alkenyl part of the enyne and that the exo-pathway was a consequence of the disfavoured steric interactions between the bulky ruthenium moiety and the sterically demanding Si(Ph)2 group in the resulting Ru-carbene intermediate.38
Recently, alkynyl silyloxy tethered enynes have been employed in a tandem CM–RCEYM sequence to form novel cyclic siloxanes.39
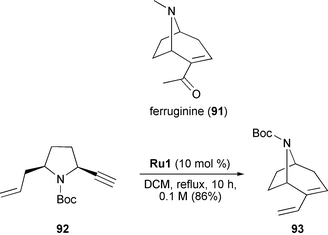
Due to its exceptional functional group and heteroatom tolerance, RCEYM has been recognized as an attractive method for the synthesis of natural products. An interesting example is the asymmetric synthesis of the tropane ferruginine (91), which was reported by Aggarwal in 2004 (Scheme 20).40 Enyne 92, prepared from L-pyroglutamic acid in 8 steps, was subjected to RCEYM in refluxing DCM with 10 mol% of Grubbs' first, second and third-generation catalysts Ru1, Ru2 and Ru3, respectively. Whereas the yields of 93 with the latter two catalysts were very low due to their high activity causing decomposition of the product, the less active first generation catalyst Ru1 proved to be more effective allowing the RCEYM to form tropane 93 in high yield (86%). Finally, three subsequent transformations, namely Wacker-oxidation, Boc-deprotection and N-methylation, led to the target compound. In summary, the synthesis of ferruginine (91) was completed in 12 steps with 29% overall yield, involving RCEYM as key transformation.
 | ||
| Scheme 20 RCEYM as key step in the synthesis of ferruginine (91). | ||
Two aspects of Aggarwal's ferruginine synthesis are particularly noteworthy. First, it is one of the few examples where enyne metathesis was employed to construct a bridged bicyclic compound, and, second, it is one of the seldom cases where Grubbs' first-generation catalyst was superior to the later generation catalysts.
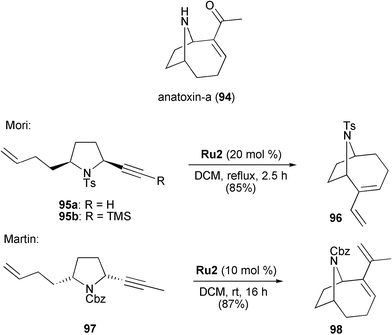
Recently, Mori41 and Martin42,43 reported (independently at almost the same time; submission dates February 27 and 29, 2004, respectively) on related syntheses of other bridged bicyclic products featuring this methodology. They utilised RCEYM reactions as key steps in their total syntheses of the potent nicotinic acetylcholine receptoragonist anatoxin-a (94) (Scheme 21).
 | ||
| Scheme 21 RCEYM reactions as key steps in syntheses of (+)-anatoxin-a (94). | ||
Mori used enynes 95 as starting material, which can be obtained from (–)-pyroglutamic acid in a few steps. Initial attempts to perform RCEYM reactions with cis-2,5-disubstituted terminal alkyne 95a and catalysts Ru1, Ru2 and Ru5 in the presence or absence of ethylene remained of only limited success, and the desired product 96 was obtained in low yield. Gratifyingly, 96 could be isolated in 85% yield when the metathesis was carried out with a combination of the silyl protected enyne 95b and 20 mol% of Grubbs' second-generation catalyst Ru2 in refluxing DCM. Four more steps from 96 were then required to finish the synthesis of anatoxin-a (94).41
Martin reported an alternative approach towards anatoxin-a (94).42,43 Starting from D-methyl pyroglutamate, enyne 97 having an internal alkynyl group was prepared in five steps. Subsequent RCEYM of 97 upon treatment with 10 mol% of Grubbs' second-generation catalyst Ru2 in DCM at room temperature proceeded smoothly to give the 9-azabicyclo[4.2.1]nonene98 in 87% yield. Three further transformations completed Martin's synthesis of anatoxin-a.
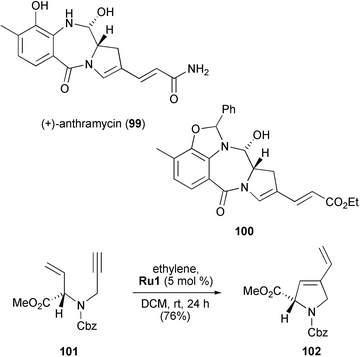
Scheme 22 shows Mori's approach towards (+)-anthramycin (99).44 Again, RCEYM was used as key method for the construction of the framework. Enyne 101, derived from L-methionine, was used in combination with 5 mol% of Grubbs' first-generation catalyst Ru1 under an ethylene atmosphere in DCM at room temperature to give pyrrolidine derivative 102 in 76% yield. Further transformations, which also include a CM reaction, led to anthramycin derivative 100, which is closely related to the target compound.
 | ||
| Scheme 22 Synthetic approach towards (+)-anthramycin (99) using RCEYM as key transformation. | ||
4. Conclusions and outlook
Enyne metathesis , and in particular, its subclass RCEYM, has become a powerful tool in organic chemistry. Although still less used than the analogous alkene metathesis , its synthetic potential has been recognized and a deeper mechanistic understanding has been achieved. This review covers both a description of recent advances in elucidating the mechanistic principles of RCEYM reactions and a presentation of selected recent applications of RCEYM reactions in the synthesis of heterocyclic compounds . In the first part, relevant pathways of RCEYM are discussed and it is shown that several factors including the nature of catalyst, the ring-size of the product, effects of ethylene and the substitution pattern of the enynes determine the mechanistic pathways. The examples in the second part reveal that RCEYM has become an impressively powerful and valuable method for the preparation of a wide range of synthetically relevant heterocycles. In particular, the high tolerance of the catalysts towards heteroatoms makes it feasible to utilise RCEYM in the construction of functionalised building blocks for target-directed natural product synthesis.Undoubtedly, the future of enyne metathesis is bright, in particular since its synthetic potential has recently been extended to tandem RCM of dienynes,45 tandem RCM–CM46 or tandem ROM–RCM reactions.47 Furthermore, alternative reactivity patterns have been discovered as exemplified by the reaction of an enyne with an alkylidene–ruthenium complex, which favours a tandem alkenylation–cyclopropanation instead of the expected RCEYM.48 We are therefore convinced that current synthetic limitations will soon be overcome and that further investigations will lead to fascinating new frontiers in enyne metathesis chemistry.
Acknowledgements
We are grateful to the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft (DFG) for financial support. Dr Ingo Schiffers is acknowledged for critical comments on the manuscript. H. V. thanks the Alexander von Humboldt-Stiftung for a postdoctoral fellowship.References
- For Nobel Prize announcement, see: Angew. Chem., Int. Ed., 2005, 44, 6982 Search PubMed.
- (a) S. T. Diver and A. J. Giessert, Chem. Rev., 2004, 104, 1317 CrossRef CAS; (b) M. Mori, J. Mol. Catal. A: Chem., 2004, 104, 1317; (c) C. S. Poulsen and R. Madsen, Synthesis, 2003, 1 CAS; (d) M. Mori, Top. Organomet. Chem., 1998, 1, 133 CAS.
- B. Schmidt, Angew. Chem., Int. Ed., 2003, 42, 4996 CrossRef CAS.
- A. Fürstner and P. W. Davies, Chem. Commun., 2005, 2307 RSC.
- D. Astruc, New J. Chem., 2005, 29, 42 RSC.
- D. E. A. Brittain, B. L. Gray and S. L. Schreiber, Chem.–Eur. J., 2005, 11, 5086 CrossRef CAS.
- For industrial applications, see: (a) T. Nicola, M. Brenner, K. Donsbach and P. Kreye, Org. Process Res. Dev., 2005, 9, 513 Search PubMed; (b) M. Poirier, N. Aubry, C. Boucher, J.-M. Ferland, S. LaPlante and Y. S. Tsantrizos, J. Org. Chem., 2005, 70, 10765 CrossRef CAS.
- (a) T. J. Katz and T. M. Sivavec, J. Am. Chem. Soc., 1985, 107, 737 CrossRef CAS; (b) T. M. Sivavec and T. J. Katz, Organometallics, 1989, 8, 1620 CrossRef CAS.
- (a) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS; (b) J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035 CrossRef CAS.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168 CrossRef CAS.
- H. Wakamatsu and S. Blechert, Angew. Chem., Int. Ed., 2002, 41, 2403 CrossRef CAS.
- A. H. Hoveyda, D. G. Gillingham, J. J. Van Veldhuizen, O. Kataoka, S. B. Garber, J. S. Kingsbury and J. P. A. Harrity, Org. Biomol. Chem., 2004, 2, 8 RSC.
- B. M. Trost and M. K. Trost, J. Am. Chem. Soc., 1991, 113, 1850 CrossRef CAS.
- B. M. Trost, M. Yanai and K. Hoogsteen, J. Am. Chem. Soc., 1993, 115, 5294 CrossRef CAS.
- L. Anorbe, G. Dominguez and J. Pérez-Castells, Chem.–Eur. J., 2004, 10, 4938 CrossRef CAS.
- M. Méndez, V. Mamane and A. Fürstner, Chemtracts, 2003, 16, 397 CAS.
- (a) A. M. Echavarren and C. Nevado, Chem. Soc. Rev., 2004, 33, 431 RSC; (b) See also in: C. Nieto-Oberhuber, S. López, E. Jiménez-Núñez and A. M. Echavarren, Chem.–Eur. J., 2006, 12, 5916 Search PubMed.
- A. Kinoshita and M. Mori, Synlett, 2004, 1020.
- S. V. Maifeld, R. L. Miller and D. Lee, J. Am. Chem. Soc., 2004, 126, 12228 CrossRef CAS.
- M. Mori, N. Sakakibara and A. Kinoshita, J. Org. Chem., 1998, 63, 6082 CrossRef CAS.
- T. R. Hoye, S. M. Donaldson and T. J. Vos, Org. Lett., 1999, 1, 277 CrossRef.
- (a) E. Vedrenne, F. Royer, J. Oble, L. El Kaïm and L. Grimaud, Synlett, 2005, 2379 CAS; (b) B. R. Galan, A. J. Giessert, J. B. Keister and S. T. Diver, J. Am. Chem. Soc., 2005, 127, 5762 CrossRef CAS.
- (a) E. C. Hansen and D. Lee, J. Am. Chem. Soc., 2003, 125, 9582 CrossRef CAS; (b) E. C. Hansen and D. Lee, J. Am. Chem. Soc., 2004, 126, 15074 CrossRef CAS; (c) For a concept article, see: S. V. Maifeld and D. Lee, Chem.–Eur. J., 2005, 11, 6118 Search PubMed.
- G. C. Lloyd-Jones, R. G. Margue and J. G. de Vries, Angew. Chem., Int. Ed., 2005, 44, 7442 CrossRef CAS.
- (a) F. Dolhem, C. Lièvre and G. Demailly, Eur. J. Org. Chem., 2003, 2336 CrossRef CAS; (b) F. Dolhem, C. Lièvre and G. Demailly, J. Org. Chem., 2004, 69, 3400 CrossRef CAS.
- (a) T. Kitamura and M. Mori, Org. Lett., 2001, 3, 1161 CrossRef CAS; (b) T. Kitamura, Y. Sato and M. Mori, Adv. Synth. Catal., 2002, 344, 678 CrossRef CAS; (c) M. Mori, H. Wakamatsu, N. Saito, Y. Sato, R. Narita, S. Sato and R. Fujita, Tetrahedron, 2006, 62, 3872 CrossRef CAS.
- N. Desroy, F. Robert-Peillard, J. Toueg, R. Duboc, C. Hénaut, M.-N. Rager, M. Savignac and J.-P. Genêt, Eur. J. Org. Chem., 2004, 4840 CrossRef CAS.
- J. Tae and D. W. Hahn, Tetrahedron Lett., 2004, 45, 3757 CrossRef CAS.
- Y. J. Kim and D. Lee, Org. Lett., 2004, 6, 4351 CrossRef CAS.
- B. G. Kim and M. L. Snapper, J. Am. Chem. Soc., 2006, 128, 52 CrossRef CAS.
- F. Monnier, D. Castillo, S. Dérien, L. Toupet and P. H. Dixneuf, Angew. Chem., Int. Ed., 2003, 42, 5474 CrossRef CAS.
- V. Gracias, A. F. Gasiecki and S. W. Djuric, Tetrahedron Lett., 2005, 46, 9049 CrossRef CAS.
- S. S. Salim, R. K. Bellingham and R. C. D. Brown, Eur. J. Org. Chem., 2004, 800 CrossRef CAS.
- J. S. Clark, F. Elustondo and M. C. Kimber, Chem. Commun., 2004, 2470 RSC.
- K. C. Majumdar, H. Rahaman, S. Muhuri and B. Roy, Synlett, 2006, 466 CrossRef CAS.
- K. C. Majumdar, H. Rahaman, R. Islam and B. Roy, Tetrahedron Lett., 2006, 47, 2111 CrossRef CAS.
- K. P. Kaliappan and V. Ravikumar, Org. Biomol. Chem., 2005, 3, 848 RSC.
- R. L. Miller, S. V. Maifeld and D. Lee, Org. Lett., 2004, 6, 2773 CrossRef CAS.
- S. Park, M. Kim and D. Lee, J. Am. Chem. Soc., 2005, 127, 9410 CrossRef CAS.
- V. K. Aggarwal, C. J. Astle and M. Roger-Evans, Org. Lett., 2004, 6, 1469 CrossRef CAS.
- M. Mori, T. Tomita, Y. Kita and T. Kitamura, Tetrahedron Lett., 2004, 45, 4397 CrossRef CAS.
- J. B. Brenneman and S. F. Martin, Org. Lett., 2004, 6, 1329 CrossRef CAS.
- J. B. Brenneman, R. Machauer and S. F. Martin, Tetrahedron, 2004, 60, 7301 CrossRef CAS.
- T. Kitamura, Y. Sato and M. Mori, Tetrahedron, 2004, 60, 9649 CrossRef CAS.
- R. Garcia-Fandiño, E. M. Codesido, E. Sobarzo-Sánchez, L. Castedo and J. R. Granja, Org. Lett., 2004, 6, 193 CrossRef CAS.
- D. A. Kummer, J. B. Brenneman and S. F. Martin, Org. Lett., 2005, 7, 4621 CrossRef CAS.
- M. Mori, Y. Kuzuba, T. Kitamura and Y. Sato, Org. Lett., 2002, 4, 3855 CrossRef CAS.
- M. Eckert, F. Monnier, G. T. Shchetnikov, I. D. Titanyuk, S. N. Osipov, L. Toupet, S. Dérien and P. H. Dixneuf, Org. Lett., 2005, 7, 3741 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |