Very large acceleration of the photoinduced electron transfer in a Ru(bpy)3–naphthalene bisimide dyad bridged on the naphthyl core†
Frédérique
Chaignon
a,
Magnus
Falkenström
b,
Susanne
Karlsson
b,
Errol
Blart
a,
Fabrice
Odobel
*a and
Leif
Hammarström
*b
aLaboratoire de Synthèse Organique, UMR 6513 CNRS & FR CNRS 2465, Université de Nantes, Faculté des Sciences et des Techniques de Nantes, BP 92208; 2, rue de la Houssinière, 44322, Nantes Cedex 03, France
bChemical Physics, Department of Photochemistry and Molecular Science, Uppsala University, Box 523, SE-751 20 Uppsala, Sweden. E-mail: Leif@fotomol.uu.se
First published on 22nd November 2006
Abstract
By linking a naphthalenebisimide (NBI) unit to [Ru(bpy)3]2+ on the naphthyl core the rate of photoinduced Ru-to-NBI electron transfer was 1000-fold increased compared to the case with a conventional linking on the nitrogen.
There is considerable interest in the development of molecular arrays for long range electron transfer in view of applications in molecular electronics and solar energy conversion.1,2 In this context, electronic communication via the bridging unit is essential for achieving fast and efficient electron transport over a long distance. Naphthalenebisimide (NBI) has many favorable properties as electron acceptor in such arrays, with a suitable reduction potential (–0.5 V vs. SCE) and a characteristic radical anion absorption spectrum .3 Moreover, a relatively high-lying triplet state (2.03 eV) makes triplet–triplet energy transfer from an appended chromophore less prone to compete with electron transfer. The latter is a particular problem for arrays based on transition metal chromophores, as has been noted in e.g. RuII(polypyridine)–fullerene arrays, where the fullerene triplet lies at only 1.25 eV.4,5 The utilization of NBI acceptors in molecular arrays has been reported previously by several groups,1,3,6,7 but the NBI was exclusively attached to the neighboring unit through the nitrogen atom of the bisimide group. We noted that the electron transfer rate has often been surprisingly slow. In e.g. a series of RuII(polypyridine)–NBI dyads7 the rate was up to three orders of magnitude slower than for corresponding dyads based on methylviologen as acceptor.8 Most of this difference can probably be attributed to a weaker electronic coupling. Indeed, the NBI frontier orbitals show a nodal plane through both the nitrogen atoms, while the main density is on the 2,3,6,7 positions of the naphthyl core.9 To increase the electronic coupling, we report herein a new type of connectivity in dyad 1 (Chart 1) by linking on the naphthyl core of NBI, which has a remarkable effect on the electron transfer rate.
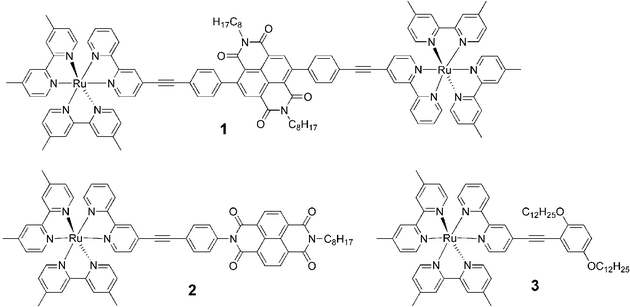 | ||
| Chart 1 Structures of the complexes. | ||
The preparation of dyads 1 and 2 is depicted in Scheme 1. Although the synthesis of dyad 2 is quite straightforward from starting materials already published in the literature, the preparation of 1 calls for the key intermediate 2,6-dibromonaphthalene tetracarboxylic dianhydride 11. The preparation of the dichloro analogue of 11 was published, but requires many steps and the utilization of noxious reagents such as chlorine gas.9,10 We found that the bromination of naphthalene tetracarboxylic dianhydride9 could be performed with dibromoisocyanuric acid10 in hot sulfuric acid solution in a quantitative yield. The poorly soluble 2,6-dibromonaphthalene-1,4:5,8-tetracarboxylic dianhydride 11 was then reacted with octylamine in acetic acid and the expected soluble N,N′-dioctyl-2,6-dibromonaphthalene-1,4,5,8-tetracarboxylic acid bisimide12 was subjected to a Stille cross-coupling reaction with an excess of 4-trimethylsilylethynyl trimethyltin benzene13 in the presence of a catalytic amount of Pd(PPh3)4 to give the bis coupled compound14 in 76% yield.
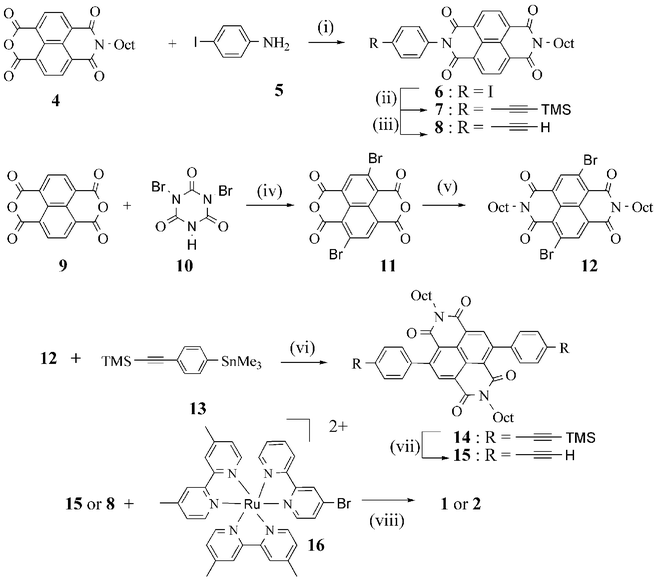 | ||
| Scheme 1 Synthetic route to prepare the dyads 1 and 2. Reagents and conditions: (i) DMF, reflux, 48%; (ii) PdCl2, Cu(OAc)2, P(Ph)3 , Et3N, THF, 70 °C, 15 h, 90%; (iii) Bu4NF, THF, R.T., 1 h, 100%; (iv) H2SO4, 130 °C, 15 h, 100%; (v) octylamine, acetic acid, 130 °C, 1 h, 28%; (vi) Pd(PPh3)4, toluene, 120 °C, 15 h, 76%; (vii) Bu4NF, THF, R.T., 1 h, 100%; (viii) Pd(dppf)Cl2, CuI, Et3N, DMF, 45–70 °C, 15 h, 40% for 1 and 47% for 2. | ||
The last step of the synthesis of 1, 2 and 3 consists of a Sonogashira cross-coupling reaction which was directly performed on the ruthenium trisbipyridine complex16 with the NBI derivatives 8 or 15. The compounds 1, 2 and 3 were thus respectively obtained with 47%, 40% and 70% yield. All the new compounds gave satisfactory 1H NMR and mass analyses.
The UV-Vis absorption spectra of the dyads 1 and 2 were simple sums of the spectra of their components: the reference ruthenium complex3 and the NBI unit (see Supporting Information). The lowest energy transition is the MLCT band of the Ru-complex around 450 nm, while the NBI unit absorption is seen around 400 nm, allowing for selective Ru-excitation. The reduction potential of the [Ru(bpy)3]3+/2+-couple, measured by cyclic voltammetry (in DMF with Bu4NPF6), was 1.21 V and 1.28 V vs. SCE, for 1 and 2 respectively, and for the NBI0/– couple it was –0.48 V and –0.56 V, respectively.
The 3MLCT emission of 3 in 298 K, deaerated acetonitrile showed a maximum at 666 nm and a lifetime of 1.5 µs. The emission intensity of the dyads was instead only 4 and 8% in 1 and 2, relative to 3. The emission lifetime for 2 was reduced to 63 ns, and in 1 it decreased dramatically to only 30 ps. Thus, the quenching rate increases by three orders of magnitude when the phenylacetylene spacer is attached to NBI through the naphthyl core compared to when it is attached through the nitrogen.
The appended NBI unit could in principle quench the 3MLCT state through both triplet energy transfer and electron transfer. Energy transfer would be isoergonic, with E00 = 2.04 eV4 for the 3MLCT state and E00 = 2.03 eV5 for 3NBI. The 3NBI lifetime is ca. 20 µs.11 Thus, if energy transfer was the main reaction one would expect an excited state equilibration and a more long-lived “delayed” Ru-emission, as opposed to the observed lifetime quenching. Instead, electron transfer to the NBI is the probable quenching mechanism, with a moderately high driving force: ΔG0 = –0.35 and –0.20 eV for 1 and 2, respectively.
For 2 the transient absorption data only showed the features of the 3MLCT decay, with the same time constant (63 ns) as obtained in the emission experiments (Supporting Information). This suggests that the electron transfer products recombine very rapidly to the ground state reactants. In 1, however, the NBI˙– radical could be traced by its absorption around 500 nm, although this is very close to the MLCT bleaching maximum, and its contribution around 605 nm (Fig. 1). The initial transient absorption (1 ps) is identical to that for the reference 3, and shows the 3MLCT state, with a bleaching maximum at 480 nm. Subsequently the isosbestic point around 500 nm blueshifts during the first tens of picoseconds, and the trace at 500 nm shows a clear rise-and-decay behavior (see inset), which is not seen in 3. This can be attributed to the NBI˙– absorption. A biexponential, global fit to the data at four different wavelengths gave τ = 52 ps for the forward electron transfer (*RuII–NBI → RuIII–NBI˙–), in fair agreement with the time resolved emission results, and τ = 14 ps for the back reaction (RuIII–NBI˙– → RuII–NBI) (Fig. 1). The short time constant for the back reaction explains the small transient signals for the charge-separated state. Nevertheless, the absorption peak around 605 nm from NBI˙– can be discerned in the spectra at 25–100 ps. A difference spectrum at 25 ps, in which the contribution of excited states remaining was subtracted, showed very clearly the features of NBI˙– (Fig. 1, right panel): absorption peaks at 474 nm (ε = 2.3 × 104 M–1 cm–1) and 605 nm (ε = 6.4 × 103 M–1 cm–1),12 while the broad RuIII–RuII bleach around 480 nm (Δε ≈ 1 × 104 M–1 cm–1) only reduced the net signal in this region. A long-lived transient with the same spectral shape as the initial 3MLCT state remained after > 1 ns and was attributed to the product of a partial photo-decomposition during measurement.
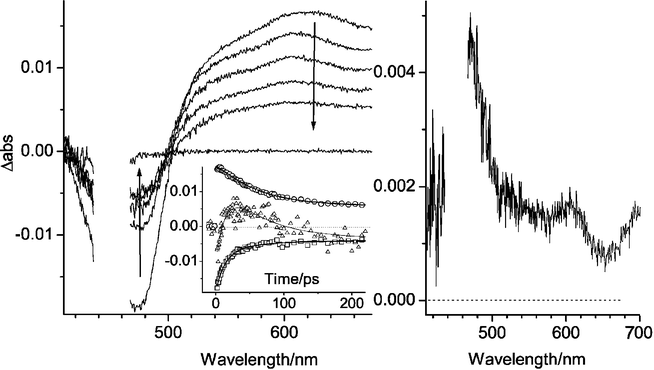 | ||
| Fig. 1 (left): Transient absorption spectra for 1 at 1 ps, 25 ps, 50 ps, 100 ps and 250 ps after excitation with a ∼ 100 fs laser pulse at 450 nm. The inset shows the kinetic traces recorded at 475 nm (squares), 500 nm (triangles; signal enlarged) and 605 nm (circles). The solid lines are biexponential fits with time constants of 14 ps and 52 ps. (right): Transient absorption spectrum after 25 ps when the contribution from the remaining excited state has been subtracted (see text); (CH3CN, 298 K). | ||
The three orders of magnitude increase in electron transfer for 1 compared to 2 shows that our design was successful. The distance between the components is nearly identical. The 0.15 eV extra driving force for electron transfer in 1 would, according to Marcus theory, only increase the rate by a factor of ca. 20.13 The main effect, accounting for the remaining two orders of magnitude increase in rate, must be attributed to the much better electronic coupling obtained by linking the spacer onto the naphthyl core. A similar enhancement of triplet energy transfer by linking a peryleneimide acceptor to the aromatic core instead of on the nitrogen has been reported.14
The quick and efficient preparation of 2,6-dibromonaphthalene-1,4,5,8-tetracarboxylic acid bisanhydride we report for the first time allows for the functionalization of NBI directly on the naphthyl core. This gives a much better electronic coupling as shown by the very large increase in rate of photoinduced electron transfer in dyad 1. This approach can be used to design new molecular systems using NBI as electron acceptor for rapid, long distance electron transfer.
We thank Prof. Dan G. Nocera (MIT) for discussions. This work was supported by the Swedish Foundation for Strategic Research, The Swedish Research Council, the Swedish Energy Agency, the European Commission (NEST-STRP “SOLAR-H”, contract no. 516510), and the French Ministry of Research for the “Action Concertée Incitative” (ACI) “Jeune Chercheur” 4057 and the ANR program “PhotoCumElec”.
Notes and references
- Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001 Search PubMed.
- J. Jortner and M. Ratner, Molecular Electronics, Blackwell Science, London, 1997 Search PubMed.
- D. Gosztola, M. P. Niemczyk, W. Svec, A. S. Lukas and M. R. Wasielewski, J. Phys. Chem. A, 2000, 104, 6545 CrossRef CAS.
- F. Chaignon, J. Torroba, E. Blart, M. Borgström, L. Hammarström and F. Odobel, New J. Chem., 2005, 29, 1272 RSC.
- N. Armaroli, G. Accorsi, D. Felder and J.-F. Nierengarten, Chem.–Eur. J., 2002, 8, 2314 CrossRef CAS; D. M. Guldi, M. Maggini, E. Menna, G. Scorrano, P. Ceroni, M. Marcaccio, F. Paolucci and S. Roffia, Chem.–Eur. J., 2001, 7, 1597 CrossRef CAS.
- N. Mataga, H. Chosrowjan, Y. Shibata, N. Yoshida, A. Osuka, T. Kikuzawa and T. Okada, J. Am. Chem. Soc., 2001, 123, 12422 CrossRef CAS; H. Shiratori, T. Ohno, K. Nozaki, I. Yamazaki, Y. Nishimura and A. Osuka, J. Org. Chem., 2000, 65, 8747 CrossRef CAS; Y. Mori, Y. Sakaguchi and H. Hayashi, J. Phys. Chem. A, 2002, 106, 4453 CrossRef CAS; K. Okamoto, Y. Mori, H. Yamada, H. Imahori and S. Fukuzumi, Chem.–Eur. J., 2004, 10, 474 CrossRef CAS; N. P. Redmore, I. V. Rubtsov and M. J. Therien, J. Am. Chem. Soc., 2003, 125, 8769 CrossRef CAS.
- D. W. Dixon, N. B. Thornton, V. Steullet and T. Netzel, Inorg. Chem., 1999, 38, 5526 CrossRef CAS; M. D. Hossain, M.-A. Haga, H. Monjushiro, B. Gholamkhass, K. Nozaki and T. Ohno, Chem. Lett., 1997, 573 CrossRef CAS; O. Johansson, M. Borgström, R. Lomoth, M. Palmblad, J. Bergquist, L. Hammarström, L. Sun and B. Åkermark, Inorg. Chem., 2003, 42, 2908 CrossRef CAS.
- E. H. Yonemoto, R. L. Riley, Y. I. Kim, S. J. Atherton, R. H. Schmehl and T. E. Mallouk, J. Am. Chem. Soc., 1992, 114, 8081 CrossRef CAS.
- F. Wurthner, S. Ahmed, C. Thalacker and T. Debaerdemaeker, Chem.–Eur. J., 2002, 8, 4742 CrossRef CAS.
- E. Bonnet, P. Gangneux and E. Marechal, Bull. Soc. Chim. Fr., 1976, 504 CAS.
- J. E. Rogers and L. A. Kelly, J. Am. Chem. Soc., 1999, 121, 3854 CrossRef CAS.
- M. J. Fuller and M. R. Wasielewski, J. Phys. Chem. B, 2001, 105, 7216 CrossRef CAS.
- Assuming that λ ≈ 1.0 eV; see R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265 Search PubMed.
- R. T. Hayes, C. J. Walsh and M. R. Wasielewski, J. Phys. Chem. A, 2004, 108, 3253 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic procedures for the preparation of new compounds, UV-Vis absorption spectra of 1–2 and transient absorption spectra for 2. See DOI: 10.1039/b615085c |
| This journal is © The Royal Society of Chemistry 2007 |