Structurally characterized intermediates in the stepwise insertion of CO–ethylene or CO–methyl acrylate into the metal–carbon bond of Pd(II) complexes stabilized by (phosphinomethyl)oxazoline ligands†
Magno
Agostinho
and
Pierre
Braunstein
*
Laboratoire de Chimie de Coordination (UMR 7177 CNRS), Institut de Chimie, Université Louis Pasteur, 4 rue Blaise Pascal, F-67070 Strasbourg Cédex, France. E-mail: braunst@chimie.u-strasbg.fr; Fax: +33 390 241 322
First published on 23rd November 2006
Abstract
The initial CO–ethylene or CO–methyl acrylate insertion steps into the Pd–Me bond of methylpalladium(II) complexes with (phosphinomethyl)oxazoline ligands, leading to metallacycles, have been fully characterized, including by X-ray diffraction.
The palladium-catalyzed alternating copolymerisation of olefins and carbon monoxide, which leads to the formation of polyketones, has become a major field of research in both academic and industrial laboratories.1
Although the basic reaction mechanism of CO–olefin copolymerisation, which involves mutually cis sites of square-planar Pd(II) species, has been established,1,2 detailed investigations on the early stages of the chain-growth process have mostly been carried out with strained alkenes owing to the difficulties often encountered in the isolation of intermediates. The first structural reports of an ethylene–CO coupling product by Green et al., using monodentate PPh3 and a N,O ligand,3a then by us using a P,O ligand3b or a diphosphine-bridged heterodimetallic Fe–Pd complex3c were followed by only a few examples with P,N,3d,e N,N3f and P,P3gchelating ligands. Furthermore, despite the considerable interest in the copolymerisation of olefins with polar monomers, such as methyl acrylate,2e,4 only a few CO–methyl acrylate coupling products have been isolated and characterized.2c,3b,c,5 Using a bidentate phosphine-imine (P,N) ligand, Reddy et al. have reported what appears to be the only structure of a CO–methyl acrylate coupling product.3e These authors used a large excess of olefin (33 to 67 equiv.) in CH2Cl2, with a reaction time between 1–3 h.
Following the synthesis of the ligands 1a,b and of the Pd(II) methyl complexes 2a,b (Scheme 1), we investigated the catalytic activity of Ni(II) complexes with 1b in ethylene oligomerisation,6 and that of the Pd(II) complexes 3a,b (Scheme 2) in ethylene–CO copolymerisation.7 Starting from 3a,b, we have now isolated the initial intermediates in CO–ethylene or CO–methyl acrylate copolymerisation reactions, without the need to use excess methyl acrylate. The structures of the new insertion products 4b, 6b and 7a,b have been determined by X-ray diffraction as well as those of the known 2a,b and 3a,b for comparative purposes.‡
 | ||
| Scheme 1 | ||
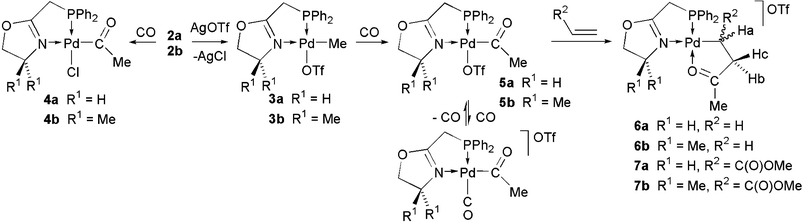 | ||
| Scheme 2 All reactions were performed at room temperature in CH2Cl2. | ||
Reactions of 2a,b and 3a,b with CO in CH2Cl2 at room temperature were monitored by 31P{1H} and 1H NMR. CO insertion into their Pd–Me bond produced within a few minutes the acyl complexes 4a,b and 5a,b, respectively (Scheme 2), as evidenced by the large high-field shift of the 31P{1H} NMR resonance (Δδ = –14.7 4a, Δδ = –14.8 4b, Δδ = –15.4 5a, Δδ = –15.1 5b, Table 1).
| IR | NMRd | |||
|---|---|---|---|---|
| ν CN | ν CO | 1H | 31P | |
| a In CH2Cl2. b In KBr, cm–1. d In CDCl3, ppm, J in Hz. | ||||
| 1a | 1660a (s) | –15.8 | ||
| 1b | 1660a (s) | –15.8 | ||
| 2a | 1647a (s) | 0.55 Pd–CH3 (d, 3JPH = 2.7) | 33.1 | |
| 2b | 1628a (s) | 0.65 Pd–CH3 (d, 3JPH = 3.3) | 32.8 | |
| 3a | 1633b (s) | 0.60 Pd–CH3 (s) | 37.4 | |
| 3b | 1632b (s) | 0.70 Pd–CH3 (s) | 37.4 | |
| 4a | 1642b (s) | 1684b (s) | 2.20 PdC(O)CH3 (d, 4JPH = 1.1) | 18.4 |
| 4b | 1632b (s) | 1685b (s) | 2.17 PdC(O)CH3 (d, 4JPH = 1.6) | 18.0 |
| 5a | 1644a (s) | 1704a (s) | 22.0 | |
| 5b | 1655a (s) | 1707a (s) | 22.3 | |
| ν CN/CO | ν C(O)OMe | |||
| 6a | 1634b (s) | 2.45 C(O)CH3 (s) | 34.4 | |
| 6b | 1629b (s) | 2.49 C(O)CH3 (s) | 34.7 | |
| 7a | 1633b (s) | 1683b (s) | 2.52 C(O)CH3 (s) | 32.8 |
| 7b | 1629b (s) | 1684b (s) | 2.55 C(O)CH3 (s) | 34.3 |
In order to detect the usually elusive palladium acyl, carbonyl complexes of the type [Pd{C(O)Me}(CO)(P,N)]OTf, a CD2Cl2 solution of 3b was exposed to an atmosphere of 13CO, and 31P{1H}, 13C{1H} and 1H NMR spectra were recorded at different temperatures. At room temperature, the 31P{1H} NMR resonance for 5b is a doublet centred at δ 21.6 (2JPC = 9.7 Hz). Its acyl carbon appears in the 13C{1H} spectrum as an intense doublet at δ 222.7 (2JPC = 9.7 Hz), and the weak signal observed at δ 179.8 corresponds to the coordinated CO of the acyl, carbonyl derivative. Upon decreasing the temperature to –60 °C, the 31P{1H} resonance significantly broadened and shifted to δ 19.1 while the 13C{1H} resonances for the acyl carbon and the coordinated CO also became broader and shifted to δ 222.6 and 175.5, respectively. This is indicative of an equilibrium between CO and OTf coordination which, at –100 °C, is completely shifted towards the acyl, carbonyl species and the 31P{1H} resonance becomes a doublet of doublets centred at δ 18.3 (2JPC(cis) = 4.1, 2JPC(trans) = 83.8 Hz) while the acyl carbon and coordinated CO13C{1H} resonances appear as doublets at δ 223.1 and 175.3, respectively (see ESI for details).† These results are in agreement with those for related complexes stabilized by P,P ligands,8 but at variance with those with a P,N ligand in which no significant amount of palladium acyl, carbonyl species was detected at –70 °C.3d
Complexes 2a,b, 3a,b and 4b have slightly distorted square planar coordination geometries with the methyl (2a,b and 3a,b) and acyl (4b) ligands cis to the phosphorus atom (ESI),† in agreement with the donor groups with the largest trans influence avoiding a mutually trans position, as observed in other complexes of the type [Pd(Me)Cl(P,N)].3e,9 The Pd–N bond distance in 2a (Table 2) is longer than that reported for the analogous PdCl2 complex [2.058(2) Å],7 which reflects the larger trans influence of the methyl group compared with chloride. The Pd–C distance of 1.9701(13) Å in 4b, although slightly shorter than in the analogous Pd–Me complex (2b), is normal for an acetyl–palladium bond. The acyl group adopts an orientation approximately perpendicular to the metal coordination plane, as observed in other complexes of the type [Pd{C(O)Me}Cl(P,N)].3e,10
| 2a | 2b | 3a | 3b | 4b | 6b | 7a | 7b | |
|---|---|---|---|---|---|---|---|---|
| Pd1–N1 | 2.103 (12) | 2.171 (3) | 2.131 (4) | 2.1550 (18) | 2.1896 (11) | 2.104 (4) | 2.082 (3) | 2.114 (3) |
| Pd1–P1 | 2.212 (4) | 2.1879 (10) | 2.1699 (12) | 2.1771 (12) | 2.2549 (13) | 2.1878 (14) | 2.2051 (10) | 2.2091 (14) |
| Pd1–C17 | 2.057 (13) | 2.049 (4) | 2.026 (5) | 2.031 (2) | 1.9701 (13) | 2.030 (5) | 2.046 (4) | 2.052 (4) |
| Pd1–O2 | 2.156 (3) | 2.1631 (17) | 2.125 (4) | 2.112 (2) | 2.138 (3) | |||
| Pd1–Cl1 | 2.383 (4) | 2.378 (1) | 2.3734 (13) | |||||
| N1–C3 | 1.291 (18) | 1.279 (5) | 1.281 (6) | 1.275 (2) | 1.2757 (15) | 1.270 (6) | 1.285 (5) | 1.273 (6) |
| C3–C4 | 1.468 (19) | 1.485 (5) | 1.476 (6) | 1.493 (3) | 1.4897 (15) | 1.480 (7) | 1.496 (5) | 1.498 (6) |
| C4–P1 | 1.847 (13) | 1.835 (4) | 1.839 (5) | 1.837 (2) | 1.8468 (12) | 1.841 (5) | 1.837 (4) | 1.835 (4) |
| C17–C18 | 1.506 (2) | 1.527 (8) | 1.545 (6) | 1.528 (7) | ||||
| C18–C19 | 1.485 (8) | 1.472 (6) | 1.486 (7) | |||||
| C19–O2 | 1.248 (6) | 1.220(5) | 1.239 (6) | |||||
| N1–Pd1–P1 | 82.2 (3) | 82.29 (9) | 84.85 (11) | 83.03 (6) | 80.66 (3) | 83.91 (12) | 82.13 (9) | 81.99 (10) |
| N1–Pd1–C17 | 176.4 (5) | 171.94 (14) | 174.97 (17) | 174.30 (8) | 173.83 (4) | 177.1 (2) | 174.17 (15) | 175.94 (15) |
| N1–Pd1–O2 | 95.14 (14) | 94.79 (7) | 100.03 (14) | 96.10 (12) | 100.26 (13) | |||
| N1–Pd1–Cl1 | 93.2 (3) | 97.94 (9) | 97.09 (3) | |||||
| P1–Pd1–O2 | 172.64 (9) | 177.32 (4) | 174.72 (10) | 175.29 (8) | 177.54 (9) | |||
| P1–Pd1–C17 | 94.5 (4) | 89.70 (13) | 90.68 (15) | 91.69 (8) | 96.35 (4) | 93.30 (16) | 99.24 (11) | 97.25 (14) |
| P1–Pd1–Cl1 | 174.66 (15) | 179.21 (4) | 173.211 (11) | |||||
| C17–Pd1–O2 | 89.00 (18) | 90.42 (8) | 82.80 (18) | 82.10 (14) | 80.57 (16) | |||
| C17–Pd1–Cl1 | 90.1 (5) | 90.09 (13) | 86.48 (4) |
Ethylene or methyl acrylate insertion into the Pd–acyl bond of 5a and 5b was completed in less than 1 h at room temperature under atmospheric pressure (31P NMR monitoring) and afforded 6a,b or 7a,b, respectively (Scheme 2). In all four complexes, coordination of the ketonic oxygen atom to Pd (see νCO, Table 1) results in a stabilizing chelate which makes β-hydrogen elimination less likely.11 These complexes are stable at room temperature for several hours in solution and weeks in the solid state, which illustrates the beneficial role of the P–N chelates . The 31P{1H} NMR signals of 6a,b and 7a,b are shifted to low field relative to those of 5a,b (Table 1). In the 1H NMR spectrum of 6a,b the Pd–CH2 protons give rise to a triplet of doublets (δ 1.65, 6a and δ 1.67, 6b) whereas the CH2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O protons appear as a broad triplet (δ 3.08, 6a and δ 3.12, 6b), indicating a smaller 4+5JHP coupling.† The CH and CH2 protons Ha, Hb and Hc of 7a,b were unambiguously identified and resonate at δ 2.46, 2.90 and 3.26 (7a) and δ 2.46, 2.87 and 3.27 (7b) respectively (vicinal and geminal JHH and JHP coupling constants are given in the ESI).†
O protons appear as a broad triplet (δ 3.08, 6a and δ 3.12, 6b), indicating a smaller 4+5JHP coupling.† The CH and CH2 protons Ha, Hb and Hc of 7a,b were unambiguously identified and resonate at δ 2.46, 2.90 and 3.26 (7a) and δ 2.46, 2.87 and 3.27 (7b) respectively (vicinal and geminal JHH and JHP coupling constants are given in the ESI).†
The crystal structures of 6b and 7a,b (see Fig. 1) were determined by X-ray diffraction and the latter two established the regioselective 2,1 insertion of methyl acrylate, which leads to an α-methoxycarbonyl complex. Deviations from idealized square planar geometries are small (Table 2). The similar Pd–C distances in 6b, 7a and 7b are in agreement with that in the only other reported structure of a CO–methyl acrylate complex.3e The Pd–O bond distances in 6b, 7a and 7b are similar and compare with those in related complexes stabilized by P,N ligands.3d,e,12 At least in the solid state, there is no interaction between the CO2Me group of 7a,b and the metal centre, which would have reduced its electrophilicity and increased its steric shielding. It is also interesting to note that related Pd(II) complexes with the P,O chelating ligand Ph2PNHC(O)Me were generally found to be less reactive than 3a,b or 5a,b, longer reaction times being required.3b
 | ||
| Fig. 1 Molecular structure of: a) complex 3b, b) the cation in 6b, c) the cation in 7b (CH groups of phenyl rings and H atoms omitted, except inserted olefin). Displacement ellipsoids are drawn at the 50% probability level. | ||
In addition to the structural characterization of CO–ethylene and CO–methyl acrylate insertion products, we have spectroscopically observed a temperature-dependent equilibrium between a triflate, acyl complex, 5b, and a cationic carbonyl, acyl Pd(II) complex. Previous studies have shown that 3a,b catalyse the CO–ethylene copolymerisation at 60–90 °C.6 Further investigations are in progress to determine the influence of the ligand bite angle on the reactivity of the chelate ring in 7a,b towards further insertion of small molecules.
We thank Luc Brissieux for preliminary results and the CNRS, the Ministère de la Recherche (Paris) and the Europeen Commission (Palladium Network HPRN-CT-2002-00196 and COST program) for support. We are grateful to Prof. R. Welter and Dr A. DeCian (ULP Strasbourg) for the crystal structure determinations and to Mrs A. Degrémont (LCC) for assistance.
Notes and references
- For recent reviews see: (a) A. Sen, Acc. Chem. Res., 1993, 26, 303 CrossRef CAS; (b) K. J. Cavell, Coord. Chem. Rev., 1996, 155, 209 CrossRef CAS; (c) E. Drent and P. H. M. Budzelaar, Chem. Rev., 1996, 96, 663 CrossRef CAS; (d) A. Sommazzi and F. Garbassi, Prog. Polym. Sci., 1997, 22, 1547 CrossRef CAS; (e) K. Nozaki and T. Hiyama, J. Organomet. Chem., 1999, 576, 248 CrossRef CAS; (f) G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428 CrossRef CAS; (g) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS; (h) G. P. Belov, Russ. Chem. Bull., Int. Ed., 2002, 51, 1605 CrossRef CAS; (i) C. Bianchini and A. Meli, Coord. Chem. Rev., 2002, 225, 35 CrossRef CAS; (j) E. Drent, J. A. M. van Broekhoven and P. H. M. Budzelaar, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 2nd edn, 2002, p. 1 Search PubMed; (k) R. A. M. Robertson and D. J. Cole-Hamilton, Coord. Chem. Rev., 2002, 225, 67 CrossRef CAS; (l) C. Bianchini, A. Meli and W. Oberhauser, Dalton Trans., 2003, 2627 RSC; (m) G. Consiglio, in Late Transition Metal Polymerization Catalysis, ed. B. Rieger, L. Saunders Baugh, S. Kacker and S. Striegler, Wiley-VCH, Weinheim, 2003, p. 279 Search PubMed; (n) J. Durand and B. Milani, Coord. Chem. Rev., 2006, 250, 542 CrossRef CAS.
- (a) J. S. Brumbaugh, R. R. Whittle, M. Parvez and A. Sen, Organometallics, 1990, 9, 1735 CrossRef CAS; (b) P. Margl and T. Ziegler, Organometallics, 1996, 15, 5519 CrossRef CAS; (c) F. C. Rix, M. Brookhart and P. S. White, J. Am. Chem. Soc., 1996, 118, 4746 CrossRef CAS; (d) M. Svensson, T. Matsubara and K. Morokuma, Organometallics, 1996, 15, 5568 CrossRef CAS; (e) S. Mecking, L. K. Johnson, L. Wang and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 888 CrossRef CAS; (f) M. A. Zuideveld, P. C. J. Kamer, P. W. N. M. van Leeuwen, P. A. A. Klusener, H. A. Stil and C. F. Roobeek, J. Am. Chem. Soc., 1998, 120, 7977 CrossRef.
- (a) M. J. Green, G. J. P. Britovsek, K. J. Cavell, B. W. Skelton and A. H. White, Chem. Commun., 1996, 1563 RSC; (b) P. Braunstein, C. Frison and X. Morise, Angew. Chem., Int. Ed., 2000, 39, 2867 CrossRef CAS; (c) P. Braunstein, J. Durand, M. Knorr and C. Strohmann, Chem. Commun., 2001, 211 RSC; (d) A. D. Burrows, M. F. Mahon and M. Varrone, Dalton Trans., 2003, 4718 RSC; (e) K. R. Reddy, K. Surekha, G.-H. Lee, S.-M. Peng, J.-T. Chen and S.-T. Liu, Organometallics, 2001, 20, 1292 CrossRef CAS; (f) S. Stoccoro, G. Minghetti, M. A. Cinellu, A. Zucca and M. Manassero, Organometallics, 2001, 20, 4111 CrossRef CAS; (g) C. Bianchini, A. Meli, W. Oberhauser, P. W. N. M. van Leeuwen, M. A. Zuideveld, Z. Freixa, P. C. J. Kamer, A. L. Spek, O. V. Gusev and A. M. Kal'sin, Organometallics, 2003, 22, 2409 CrossRef CAS.
- (a) L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267 CrossRef CAS; (b) E. Drent, R. van Dijk, R. van Ginkel, B. van Oort and R. I. Pugh, Chem. Commun., 2002, 744 RSC; (c) T. Kochi, K. Yoshimura and K. Nozaki, Dalton Trans., 2005, 25 RSC.
- (a) F. Ozawa, T. Hayashi, H. Koide and A. Yamamoto, J. Chem. Soc., Chem. Commun., 1991, 1469 RSC; (b) G. P. C. M. Dekker, C. J. Elsevier, K. Vrieze, P. W. N. M. van Leeuwen and C. F. Roobeek, J. Organomet. Chem., 1992, 430, 357 CrossRef CAS; (c) F. C. Rix and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 1137 CrossRef CAS.
- (a) F. Speiser, P. Braunstein, L. Saussine and R. Welter, Organometallics, 2004, 23, 2613 CrossRef CAS; (b) F. Speiser, P. Braunstein and L. Saussine, Acc. Chem. Res., 2005, 38, 784 CrossRef CAS.
- P. Braunstein, M. D. Fryzuk, M. Le Dall, F. Naud, S. J. Rettig and F. Speiser, J. Chem. Soc., Dalton Trans., 2000, 1067 RSC.
- (a) J. Ledford, C. S. Shultz, D. P. Gates, P. S. White, J. M. DeSimone and M. Brookhart, Organometallics, 2001, 20, 5266 CrossRef CAS; (b) J. Liu, B. T. Heaton, J. A. Iggo, R. Whyman, J. F. Bickley and A. Steiner, Chem.–Eur. J., 2006, 12, 4417 CrossRef CAS.
- (a) M. Agostinho, A. Banu, P. Braunstein, R. Welter and X. Morise, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m81 CrossRef; (b) A. Apfelbacher, P. Braunstein, L. Brissieux and R. Welter, Dalton Trans., 2003, 1669 RSC.
- (a) R. E. Ruelke, V. E. Kaasjager, P. Wehman, C. J. Elsevier, P. W. N. M. van Leeuwen, K. Vrieze, J. Fraanje, K. Goubitz and A. L. Spek, Organometallics, 1996, 15, 3022 CrossRef; (b) K. R. Reddy, W.-W. Tsai, K. Surekha, G.-H. Lee, S.-M. Peng, J.-T. Chen and S.-T. Liu, J. Chem. Soc., Dalton Trans., 2002, 1776 RSC.
- (a) J. X. McDermott, J. F. White and G. M. Whitesides, J. Am. Chem. Soc., 1973, 95, 4451 CrossRef CAS; (b) J. X. McDermott, J. F. White and G. M. Whitesides, J. Am. Chem. Soc., 1976, 98, 6521 CrossRef CAS.
- K. R. Reddy, C.-L. Chen, Y.-H. Liu, S.-M. Peng, J.-T. Chen and S.-T. Liu, Organometallics, 1999, 18, 2574 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Complete sets of crystallographic parameters for 2a,b, 3a,b, 4b, 6b, 7a,b; experimental procedures and spectroscopic characterizations. See DOI: 10.1039/b613865a |
| ‡ CCDC 622170–622177. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b613865a |
| This journal is © The Royal Society of Chemistry 2007 |