Metal cages using a bulky phosphonate as a ligand†
Viswanathan
Baskar
*,
Muralidharan
Shanmugam
,
E. Carolina
Sañudo
,
Maheswaran
Shanmugam
,
David
Collison
,
Eric J. L.
McInnes
,
Qiang
Wei
and
Richard E. P.
Winpenny
*
Department of Chemistry, The University of Manchester, Oxford Road, Manchester, UK M13 9PL. E-mail: richard.winpenny@man.ac.uk
First published on 23rd November 2006
Abstract
The synthesis, structure, magnetic and electronic properties of soluble transition metal phosphonate cages utilizing tritylphosphonic acid (TPA) as ligand are reported.
Multinuclear transition metal cages are heavily studied for several reasons including synthesis of single molecule magnets1 and to prepare model compounds to understand the structure and spectral properties of the active sites in proteins and enzymes responsible for carrying out vital chemical transformations.2 For example, the crystal structure elucidation of Synechococcus elongates PS II at 3.5 Å resolution3 suggests that the water oxidase active site consists of a Mn3O4Ca cubane core with a fourth manganese atom attached through one of the corner oxygens of the cube.4 In the same vein, copper-mediated activation of oxygen involves enzymes such as ascorbate oxidase which contain a tri-copper active site with another mononuclear “blue” copper at a distance of 12 Å.5
Here, we report the use of a bulky phosphonate (TPA)6 for synthesizing tetranuclear cages of Mn, Co and Cu. The most commonly encountered difficulty with phosphonates as ligands is the insolubility they impart to metal cages, often leading to the isolation of intractable materials. Use of solvothermal methods or co-ligands has led to multimetallic phosphonate cages,7 and we have reported the synthesis of manganese and iron cages using preformed metal carboxylate triangles as starting materials.8 Despite these developments, transition metal phosphonate cages remain comparatively rare. Hence, we were interested in further routes to discrete metal phosphonate cages and one idea was that increasing the steric bulk of the organic group bound to the phosphorus atom might restrict oligomerisation, leading to the isolation of discrete cages.
Hydrated metal(II) acetates of manganese, cobalt and copper were stirred independently with TPA in acetonitrile for 6 h at room temperature in the presence of small amounts of pyridine as a base.‡ Crystallization was carried out either by slow evaporation of acetonitrile solutions at room temperature or by diffusion methods (DCM–hexane in ratio 1 : 1). Single crystal X-ray analysis revealed the formation of [Mn4(O)(Ph3CPO3)4Py4] 1, [Co4(Ph3CPO3)4Py4] 2 and [Cu4(OH)(Ph3CPO3)3(Ph3CPO2OH)Py4·H2O·CH3CN] 3.
Structural characterization of 1 reveals the formation of a tetranuclear manganese cage (Fig. 1). It can be described as a trimer–monomer cage consisting of a µ3-oxo-centred triangle with the fourth Mn atom residing on top of the triangular unit giving an elongated tetrahedron. Four dianionic phosphonate ligands are present in the cage binding the metals in [3.111] bridging mode (Harris notation).9 One TPA coordinates exclusively to the oxo-centred triangle while the other three ligands bind the triangle to the fourth metal present. Each metal is coordinated to a pyridine molecule to complete its coordination sphere. The metals in the triangular unit have a trigonal bipyramidal geometry and the fourth metal has a distorted tetrahedral arrangement.10 Charge balance and bond valence sum calculation11 suggests that 1 is a mixed valent cage with two manganese (in the triangle) in the +3 oxidation state while the other two Mn atoms are in the +2 state. The Mn–Mn distances vary from 3.19 Å to 3.88 Å. The distances between the µ3-oxygen and Mn atoms in the triangle are 1.87 Å, 1.88 Å and 2.41 Å. The fourth Mn lies at a distance of 2.71 Å from the µ3-oxygen.
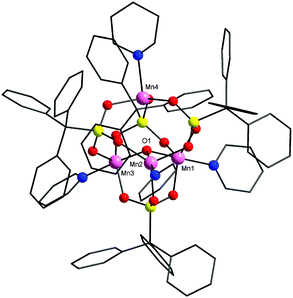 | ||
| Fig. 1 The structure of 1 in the crystal. Pink, Mn; red, O; yellow, P; blue, N; black, C. H-atoms omitted for clarity. | ||
2 is a regular tetrahedral cobalt cage (Fig. 2). All the cobalt atoms are in the divalent oxidation state (BVS calculation). The coordination environment around the metal centres is tetrahedral with each metal binding to three oxygens from different phosphonate ligands and a pyridyl nitrogen. The phosphonates again show the [3.111] bridging mode and lie on the four triangular faces. The cage diameter (distance between Co and P atom situated at opposite edges of a cube) is 5.4 Å approximately. Similar cage type structures have been reported with main-group metal phosphonates12 whereas with transition metals they are unknown.
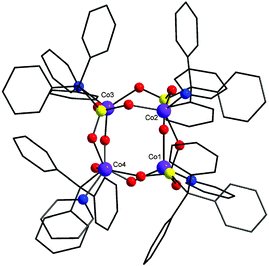 | ||
| Fig. 2 The structure of 2 in the crystal. Colours as Fig. 1 plus violet, Co. | ||
The structure of 3 shows the formation of a tetranuclear copper cage (Fig. 3). It can also be described as a trimer–monomer structure similar to 1 but for 3 the binding modes of TPA and geometry of the metal centres differ. The cage core consists of a µ3-hydroxide centred triangular unit with the fourth copper present on top of the triangle. The phosphonates show both [3.111] and [2.110] bridging modes. The two phosphonates showing the [2.110] bridging mode have a strong O…H–O H-bond between them (O–O separation = 2.40 Å).
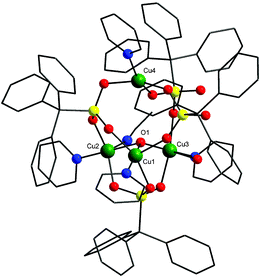 | ||
| Fig. 3 The structure of 3 in the crystal. Colours as Fig. 1 plus green, Cu. | ||
All the three coppers present in the triangle are five coordinate with distorted square pyramidal geometries while the final metal, Cu(4), is four coordinate with a a severely distorted square planar geometry (the trans angles, O–Cu–O and O–Cu–N are 158°).
All of the copper atoms in the triangle are bound to the µ3-hydroxide and to O-atoms from a 3.111-bound phosphonate. Two are also bound to one further TPA-oxygen, a terminal pyridine and a solvent molecule (either water or MeCN). The third copper binds to two phosphonate O-atoms and a pyridyl nitrogen. The inter copper distances in the triangular unit are 3.11, 3.26 and 3.61 Å, with the fourth copper 3.93, 4.97 and 5.19 Å from the three Cu centres of the triangle.
The magnetic and EPR behaviour of 1 and 2 show anti-ferromagnetic (AF) exchange between the metal centres and are uninteresting (see supplementary material). The low temperature EPR spectra of 3 at both K- and Q-band are remarkable with multiple features that are difficult to explain. The second derivative of the experimental spectrum recorded at Q-band with its simulation is shown in Fig. 4. Beginning at low field we see a multiplet pattern centred at g = 2.32 which contains more than seven lines due to copper hyperfine coupling. The next clear feature is a hyperfine quartet at g = 2.17; there are weaker features between these two multiplets. There are then four further higher field features between g = 2.0 and 2.1 without hyperfine structure. These high field features do not occur at precisely the same g-values at K- and Q-band. These spectra have the appearance of S = ½ states which is unexpected given that four S = 1/2 centres should generate integer spin states for the entire cage . However, weak features at low field are observed in S-band spectra, which could be spin forbidden transitions of an integer spin state. This spectroscopic behaviour, S = ½ like spectra with weakly frequency dependent g-values and forbidden transitions only observable at low-frequency/field, is consistent with the exchange coupling of the fourth copper to the triangle being very weak – of similar magnitude to or smaller than the Zeeman interaction. Such spectra are reminiscent of the EPR spectroscopy of multi-copper oxidases.5
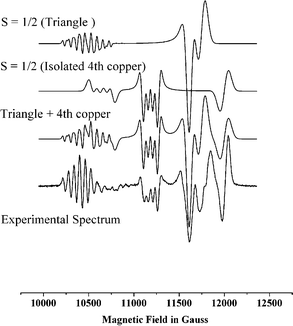 | ||
| Fig. 4 Experimental EPR spectrum of 3 at 10 K at 35 GHz (second derivative) and simulation using parameters given in the text. | ||
The structure suggests that the copper centres in the triangle should be strongly coupled to one another as they are linked by a single atom bridge, O(1). Hence, we start our interpretation of the EPR with a simplified model assuming two independent paramagnetic species – the triangle and the fourth copper [Cu(4)]. Coupling three S = 1/2 ions, S1, S2 and S3 in a triangle generates three total spins |0, 1/2>, |1, 1/2> and |1, 3/2> in the |S*, ST> notation, where S* is the intermediate spin from coupling S1 and S2, and ST is the total spin of the triangle. For a scalene triangle with anti-ferromagnetic exchange one of the ST = 1/2 states will be the ground state , with ST = 3/2 at highest energy. Assuming the strong exchange limit Bencini and Gatteschi13 have calculated that the hyperfine pattern for the |0, 1/2> and |1, 1/2> states should be very different. For |0, 1/2> the projection coefficients of the individual coppers to the total spin are c1 = c2 = 0; c3 = 1, i.e. the observed hyperfine is a quartet arising from only one copper (S3) with magnitude equal to that of the single ion. For |1, 1/2> the projection coefficients are c1 = c2 = 2/3; c3 = –1/3, i.e. we will see hyperfine to all three copper ions, where the coupling to two spins (S1 and S2) is twice that to the third, and two-thirds that of the single ion value (assuming all three spins have the same single ion value, which need not be the case for the distorted geometries observed for 3). Comparing this analysis with the spectra we have assigned the multiplet at g = 2.32 to the parallel component of the |1, 1/2> state and simulated this multiplet with |ACu1| = |ACu2| = 65 G, |ACu3| = 40 G which gives an eight line pattern which is very sensitive to the ACu1,2/ACu3 ratio. Similar multiplet structure arising from the anti-ferromagnetic coupling of three copper ions has been observed.14
The fourth copper [Cu(4)] is attached to this triangle only through phosphonate ligands, which are expected to provide a poor superexchange path. Clearly an isolated copper would give a spectrum due to an S = 1/2 state. Therefore the quartet at g = 2.17 with an observed hyperfine coupling of 50 G must be either due to this “isolated” copper or to the |0, 1/2> state of the triangle. The lineshape is not consistent with a simple parallel feature.
The four high field features are presumably the “perpendicular” resonances of the rhombic S = 1/2 spectra. Given the weak frequency dependence of their g-values and the observation of very weak forbidden transitions at low field/frequency, this assignment is clearly an oversimplification and full assignment and simulation of these spectra will require a single crystal study and the introduction of a very weak exchange between Cu(4) and the triangle; this is in progress.
We expect this exchange interaction to be of the same order as the Zeeman interaction. However a problem arises from fitting magnetic susceptibility data for 3 (see supplementary material). We have fitted the data with two Hamiltonians: both fits are reasonable and give, with g = 2.153: A, J1 = 100, J2 = 40, J3 = 12 cm–1; B, J1 = 80, J2 = 55, J3 = 27, J4 = 11 cm–1. The size of the coupling between the triangle and Cu(4) is not consistent with our interpretation of the EPR spectra, however the significant difference between these sets of J-values implies that a wide range of parameters could be used to fit to the susceptibility data well.
| H = J1SCu1SCu3 + J2(SCu1SCu2 + SCu2SCu3) + J3(SCu1SCu4 + SCu2SCu4 + SCu3SCu4) | (A) |
| H = J1SCu1SCu3 + J2(SCu1SCu2 + SCu2SCu3) + J3(SCu1SCu4) + J4(SCu2SCu4 + SCu3SCu4) | (B) |
While more work is needed to understand the spectroscopy, we believe that we understand the synthesis: the steric bulk of the large phosphonate requires the metal sites to be as far apart as possible, and this is best achieved by having a pseudo-tetrahedral cage . Alternative arrays for {M4} cages, such as a “butterfly” would be impossible with such large phosphonates. Thus we are achieving a degree of control over reactivity by using these ligands.
This work was made possible by a Royal Society Anglo-Indian Fellowship (to V. B.) and supported by the EPSRC (UK).
Notes and references
- (a) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS; (b) D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268 CrossRef CAS and references therein; (c) A. K. Boudalis, C. P. Raptopoulou, B. Abarca, R. Ballesteros, M. Chadlaoui, J. P. Tuchagues and A. Terzis, Angew. Chem., Int. Ed., 2006, 45, 432 CrossRef CAS.
- (a) S. Mukhopadhyay, S. K. Mandal, S. Bhaduri and W. H. Armstrong, Chem. Rev., 2004, 104, 3981 CrossRef CAS; (b) E. I. Solomon, M. J. Baldwin and M. D. Lowery, Chem. Rev., 1992, 92, 521 CrossRef CAS; (c) N. Kitajima and Y. Moro-oka, Chem. Rev., 1994, 94, 737 CrossRef CAS; (d) I. Bento, L. O. Martins, G. G. Lopes, M. A. Carrondo and P. F. Lindley, Dalton Trans., 2005, 3507 RSC.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831 CrossRef CAS.
- A. Mishra, W. Wernsdorfer, K. A. Abboud and G. Christou, Chem. Commun., 2005, 1, 54 RSC.
- (a) A. Messerschmidt, A. Rossi, R. Ladenstein, R. Huber, M. Bolognesi, G. Gatti, A. Marchesini, R. Petruzzelli and A. Finazzi-Agro, J. Mol. Biol., 1989, 206, 513 CrossRef CAS; (b) A. P. Cole, D. E. Root, P. Mukherjee, E. I. Solomen and T. D. P. Stack, Science, 2005, 273, 1848; (c) E. I. Solomon and M. D. Lowery, Science, 1993, 259, 1575 CAS.
- D. R. Boyd and G. Chignell, J. Chem. Soc., Trans., 1923, 123, 813 RSC.
- (a) V. Chandrasekhar and S. Kingsley, Angew. Chem., Int. Ed., 2000, 39, 2320 CrossRef CAS; (b) V. Chandrasekhar, L. Nagarajan, K. Gopal, V. Baskar and P. Koegerler, Dalton Trans., 2005, 41, 3143 RSC; (c) V. Chandrasekhar, P. Sasikumar, R. Boomishankar and G. Anantharaman, Inorg. Chem., 2006, 45, 3344 CrossRef CAS; (d) H. C. Yao, Y. Z. Li, Y. Song, Y. S. Ma, L. M. Zheng and X. Q. Xin, Inorg. Chem., 2006, 45, 59 CrossRef CAS.
- (a) E. I. Tolis, M. Helliwell, S. Langley, J. Raftery and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2003, 42, 3804 CrossRef; (b) S. Maheswaran, G. Chastanet, S. J. Teat, T. Mallah, R. Sessoli, W. Wernsdorfer and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2005, 44, 5044 CrossRef CAS.
- R. A. Coxall, S. G. Harris, D. K. Henderson, S. Parsons, P. A. Tasker and R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2000, 2349 RSC.
- T. Afrati, C. Dendrinou-Samara, C. P. Raptopoulou, A. Terzis, V. Tangoulis and D. P. Kessissoglou, Angew. Chem., Int. Ed., 2002, 41, 2148 CrossRef CAS.
- (a) I. D. Brown and K. K. Wu, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 1957 CrossRef; (b) W. Liu and H. H. Thorp, Inorg. Chem., 1993, 32, 4102 CrossRef CAS.
- (a) M. Walawalker, H. W. Roesky and R. Murugevel, Acc. Chem. Res., 1999, 32, 117 CrossRef CAS; (b) M. Walawalker, R. Murugevel, H. W. Roesky and H. G. Schmidt, Inorg. Chem., 1997, 36, 4202 CrossRef CAS.
- A. Bencini and D. Gatteschi, EPR Spectroscopy of Exchange Coupled Systems, Springer Verlag, Berlin, 1989 Search PubMed.
- X. Liu, J. A. McAllister, M. P. de Miranda, E. J. L. McInnes, C. A. Kilner and M. A. Halcrow, Chem.–Eur. J., 2004, 10, 1827–1837 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/b613756c |
| ‡ 1. Mn(OAc)2·4H2O (0.22 g, 0.9 mmol) and TPA (0.30 g, 0.9 mmol) were dissolved in CH3CN (15 ml). Pyridine (2 ml) was added to the reaction mixture, which was stirred at room temperature for 6 h and filtered. Slow evaporation of the filtrate at room temperature yielded brown crystals of 1 after a week. Yield: 0.12 g (33%). Elemental analysis for 1 (%): Calcd. C 62.61, H 4.38, N 3.04; found C 61.71, H 4.28, N 2.92%. 2. Co(OAc)2·6H2O (0.25 g, 1.0 mmol) and TPA (0.32 g, 1.0 mmol) were dissolved in CH3CN (15 ml). Pyridine (2 ml) was added to the reaction mixture, which was stirred at room temperature for 6 h and filtered. Evaporation of the filtrate afforded a blue solid, which was dissolved in DCM. Blue crystals of 2 were grown by a diffusion method (DCM–hexane). Yield: 0.37 g (80%). Elemental analysis for 2 (%): Calcd. C 62.61, H 4.38, N 3.04; found C 62.21, H 4.30, N 3.02%. 3. Cu(OAc)2·H2O (0.14 g, 0.7 mmol) and TPA (0.25 g, 0.7 mmol) were taken in CH3CN (15 ml) and the same procedure as given for 2 was followed to afford green crystals of 3. Yield: 0.21 g (57%). Elemental analysis for 3 (%): Calcd. C 60.83, H 4.43, N 3.61; found C 60.95, H 4.58, N 3.85%. Crystal data: 1: a = 16.1648(11), b = 17.4547(15), c = 19.2761(13) Å, α = 79.700(6), β = 75.423(6), γ = 69.810(7)°, V = 4914.9(6) Å3, M = 2040.04, R1 = 0.1021; 2: a = 14.8919(10), b = 34.184(2), c = 18.7673(13) Å, β = 100.7460(10)°, V = 9386.3(11) Å3, M = 2074.79, R1 = 0.0475; 3: a = 13.620(4), b = 17.079(5), c = 23.954(6) Å, α = 71.616(5), β = 73.921(6), γ = 68.045(5)°, V = 4823(2) Å3, M = 2043.20, R1 = 0.0719. Data collection, structure solution and refinement used SHELXL. CCDC 622125–622127. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b613756c |
| This journal is © The Royal Society of Chemistry 2007 |